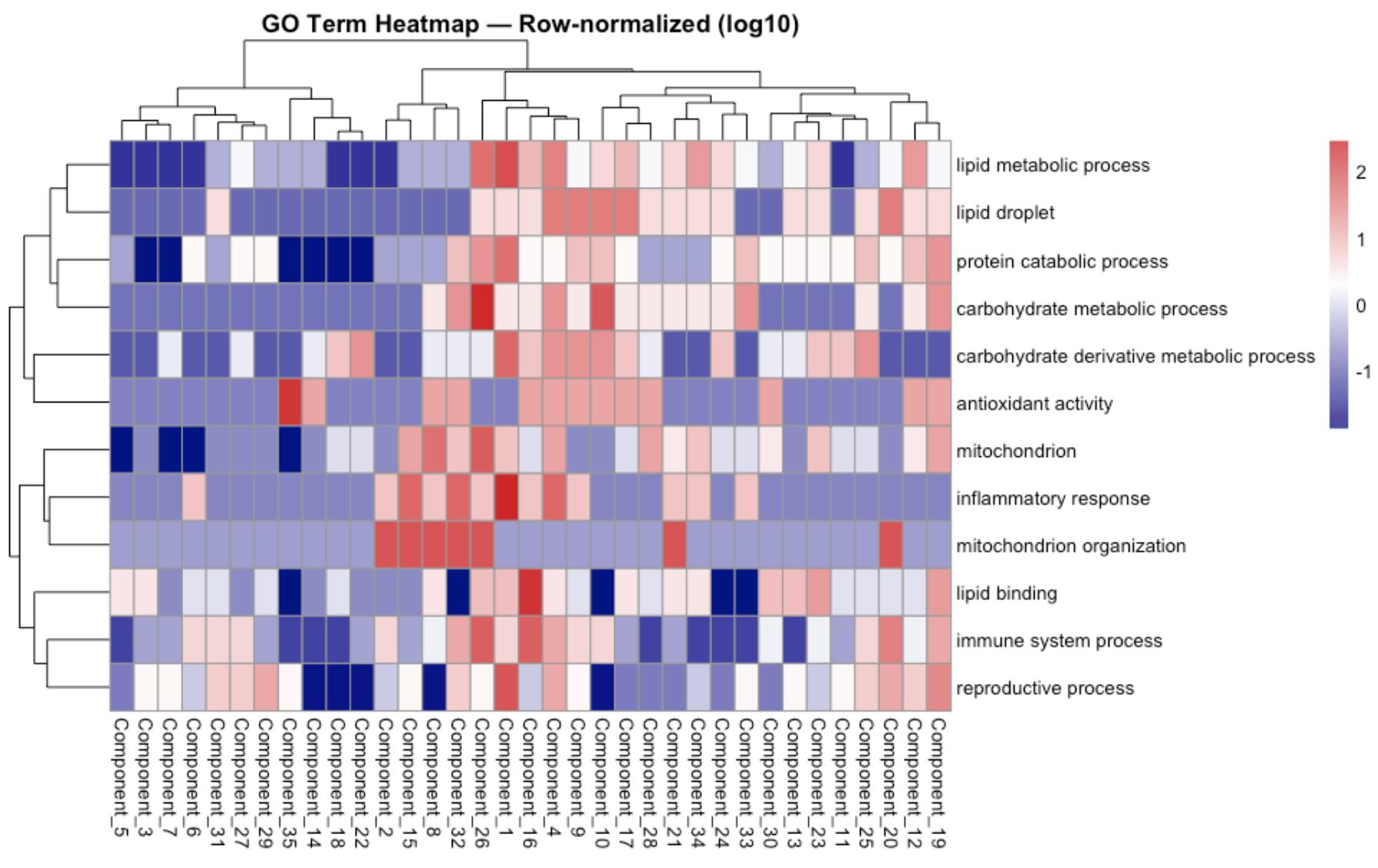
Based on overlap of genes across components wanted to increase sparsity be increasing lambda-gene values..
Ran with multiple values
LAMBDAS_GENE="0.00 0.01 0.05 0.1 0.2 0.4 0.8"
LAMBDA_SAMPLE=0.1
LAMBDA_TIME=0.05
RANK=35
OUTDIR_BASE=../output/26-rank35-optimization
for LG in $LAMBDAS_GENE; do
OUTDIR=${OUTDIR_BASE}/lambda_gene_${LG}
mkdir -p "$OUTDIR"
/Users/sr320/.local/bin/uv run python ../scripts/14.1-barnacle/build_tensor_and_run.py \
--input-file ../output/14-pca-orthologs/vst_counts_matrix.csv \
--output-dir "$OUTDIR" \
--rank $RANK \
--lambda-gene $LG \
--lambda-sample $LAMBDA_SAMPLE \
--lambda-time $LAMBDA_TIME \
--max-iter 2000 \
--tol 1e-5 \
--seed 81
done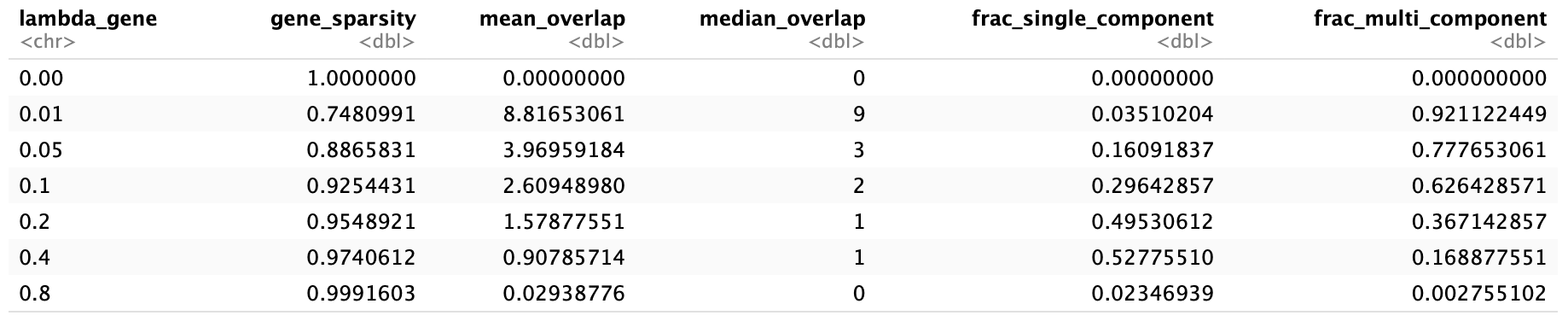
Move forward with lamda_gene of 0.2…
Sample

Time
library(tidyverse)
# File path
file_path <- "../output/26-rank35-optimization/lambda_gene_0.2/barnacle_factors/time_factors.csv"
# Read data
df <- read_csv(file_path)
# Compute variance of each component across timepoints
var_components <- df %>%
pivot_longer(cols = starts_with("Component_"),
names_to = "Component",
values_to = "Value") %>%
group_by(Component) %>%
summarise(Variance = var(Value, na.rm = TRUE)) %>%
arrange(desc(Variance))
# Select top 10 most variable components
top10_components <- var_components %>% slice_max(Variance, n = 10) %>% pull(Component)
# Prepare data for plotting
df_long <- df %>%
pivot_longer(cols = starts_with("Component_"),
names_to = "Component",
values_to = "Value") %>%
filter(Component %in% top10_components)
# Line plot
ggplot(df_long, aes(x = OG_ID, y = Value, group = Component, color = Component)) +
geom_line(linewidth = 1.2) +
geom_point(size = 2) +
theme_minimal(base_size = 14) +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
labs(
title = "Top 10 Most Variable Components Across Timepoints",
x = "Time Point",
y = "Component Value"
)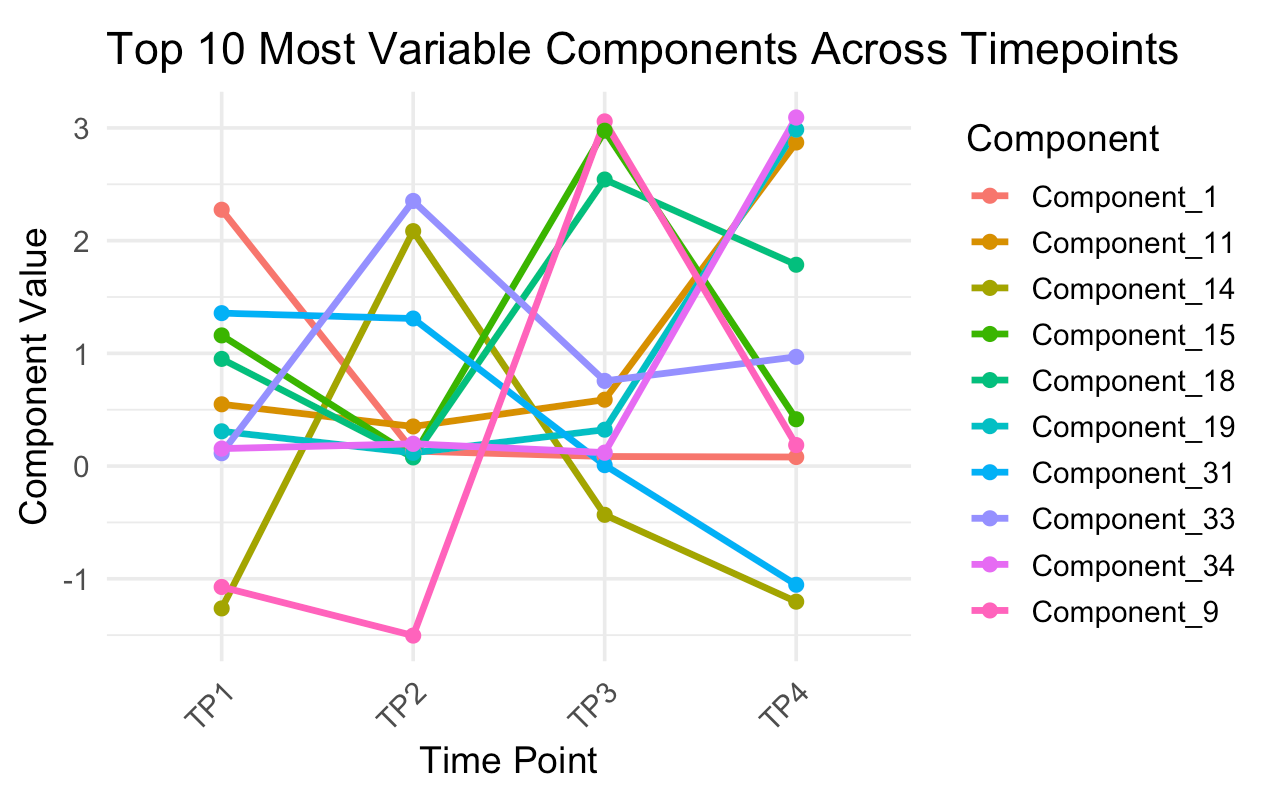
Pick your component
library(tidyverse)
# File path
file_path <- "../output/26-rank35-optimization/lambda_gene_0.2/barnacle_factors/time_factors.csv"
# ---- USER INPUT ----
# Pick the component you want to plot (e.g., "Component_5")
selected_component <- "Component_24"
# ---------------------
# Read data
df <- read_csv(file_path)
# Convert to long format
df_long <- df %>%
pivot_longer(
cols = starts_with("Component_"),
names_to = "Component",
values_to = "Value"
)
# Check that the chosen component exists
if (!(selected_component %in% unique(df_long$Component))) {
stop(paste("Component", selected_component, "not found in data!"))
}
# Filter for the selected component
df_sel <- df_long %>% filter(Component == selected_component)
# Plot single component across timepoints
ggplot(df_sel, aes(x = OG_ID, y = Value, group = 1)) +
geom_line(linewidth = 1.2, color = "steelblue") +
geom_point(size = 3, color = "firebrick") +
theme_minimal(base_size = 14) +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
labs(
title = paste0(selected_component, " Across Timepoints"),
x = "Time Point",
y = "Loading Value"
)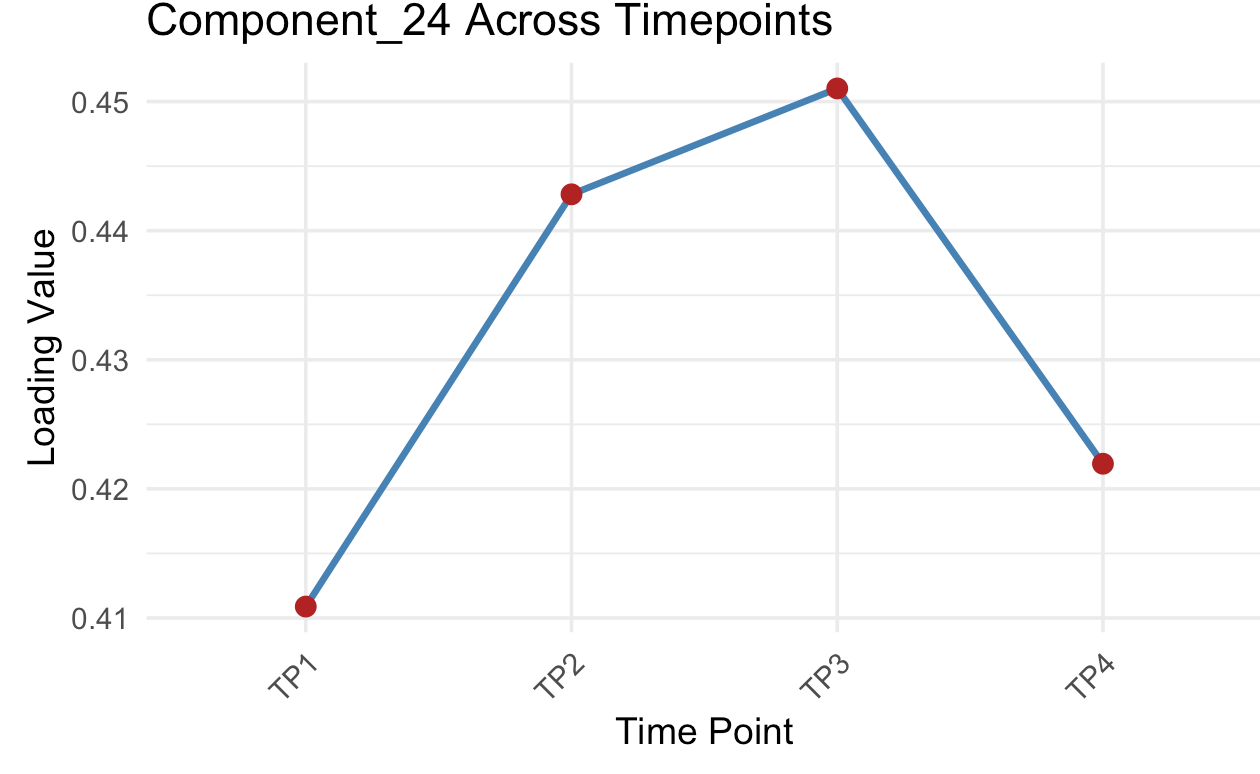
Gene Factor Loading

Annotation
An Annotation - U pick!
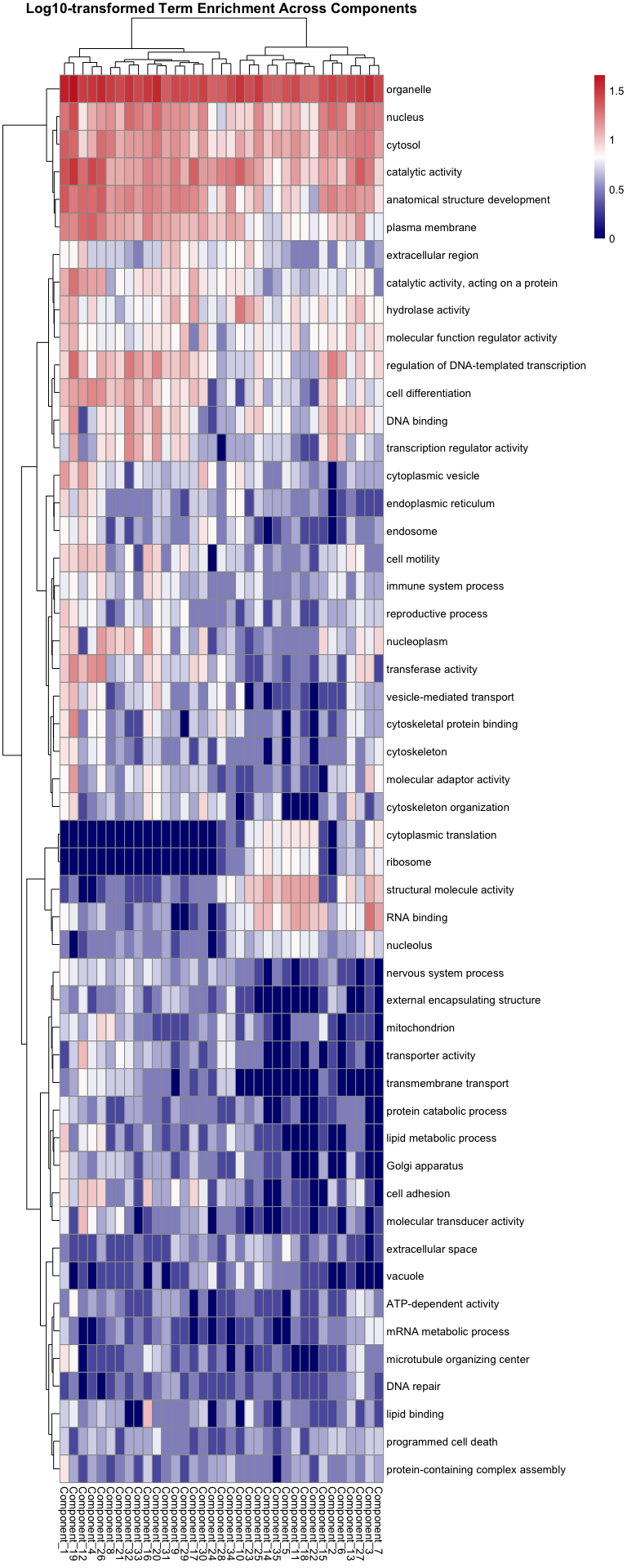
library(tidyverse)
library(pheatmap)
# ---- File path ----
file_path <- "../output/26-rank35-optimization/lambda_gene_0.2/top_genes_per_component/goslim_term_counts.csv" # change to your actual CSV path
# ---- User input ----
# Enter one or more GO terms (exact or partial match)
selected_terms <- c("protein catabolic process", "lipid", "immune system process", "carbohydrate", "antioxidant activity", "mitochondrion", "inflammatory response", "reproductive") # example
# ---- Options ----
row_normalize <- TRUE # TRUE for row-normalized (z-score), FALSE for raw
log_transform <- TRUE # TRUE to apply log10(x+1) before plotting
# ---- Load and prepare data ----
df <- read_csv(file_path)
# Remove total column if present
df <- df %>% select(-any_of("total"))
# Filter by GO term (partial match supported)
df_sel <- df %>%
filter(if_any(term, ~ str_detect(.x, paste(selected_terms, collapse = "|"))))
if (nrow(df_sel) == 0) {
stop("⚠️ No matching GO terms found. Check spelling or capitalization.")
}
# Convert to matrix
mat <- df_sel %>%
column_to_rownames(var = "term") %>%
as.matrix()
# Optional log-transform
if (log_transform) {
mat <- log10(mat + 1)
}
# Optional row normalization
if (row_normalize) {
mat <- t(scale(t(mat))) # z-score per row
}
# ---- Plot heatmap ----
pheatmap(
mat,
scale = "none",
color = colorRampPalette(c("navy", "white", "firebrick3"))(200),
clustering_method = "ward.D2",
show_rownames = TRUE,
main = paste(
"GO Term Heatmap —",
if (row_normalize) "Row-normalized" else "Raw values",
if (log_transform) "(log10)" else ""
)
)