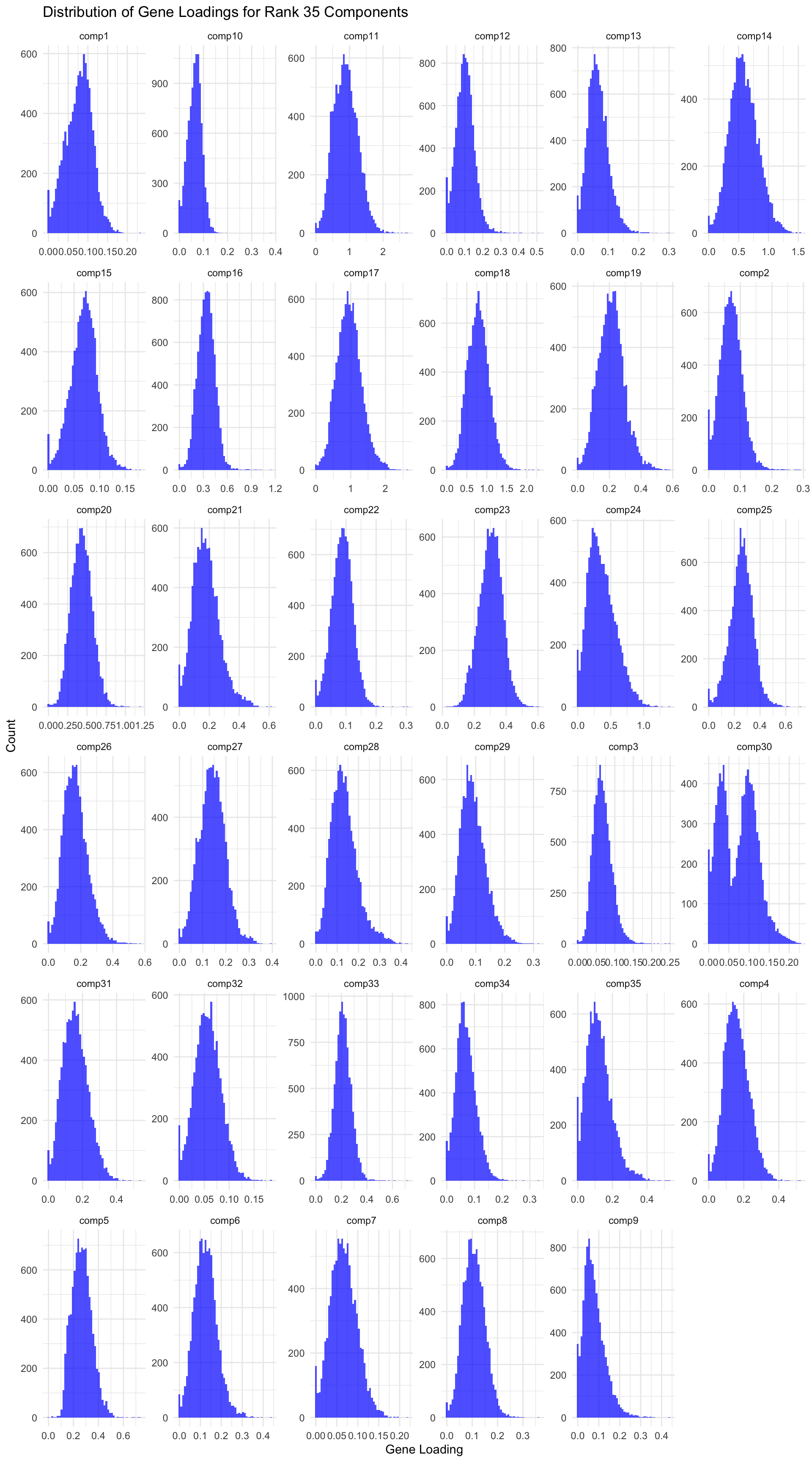
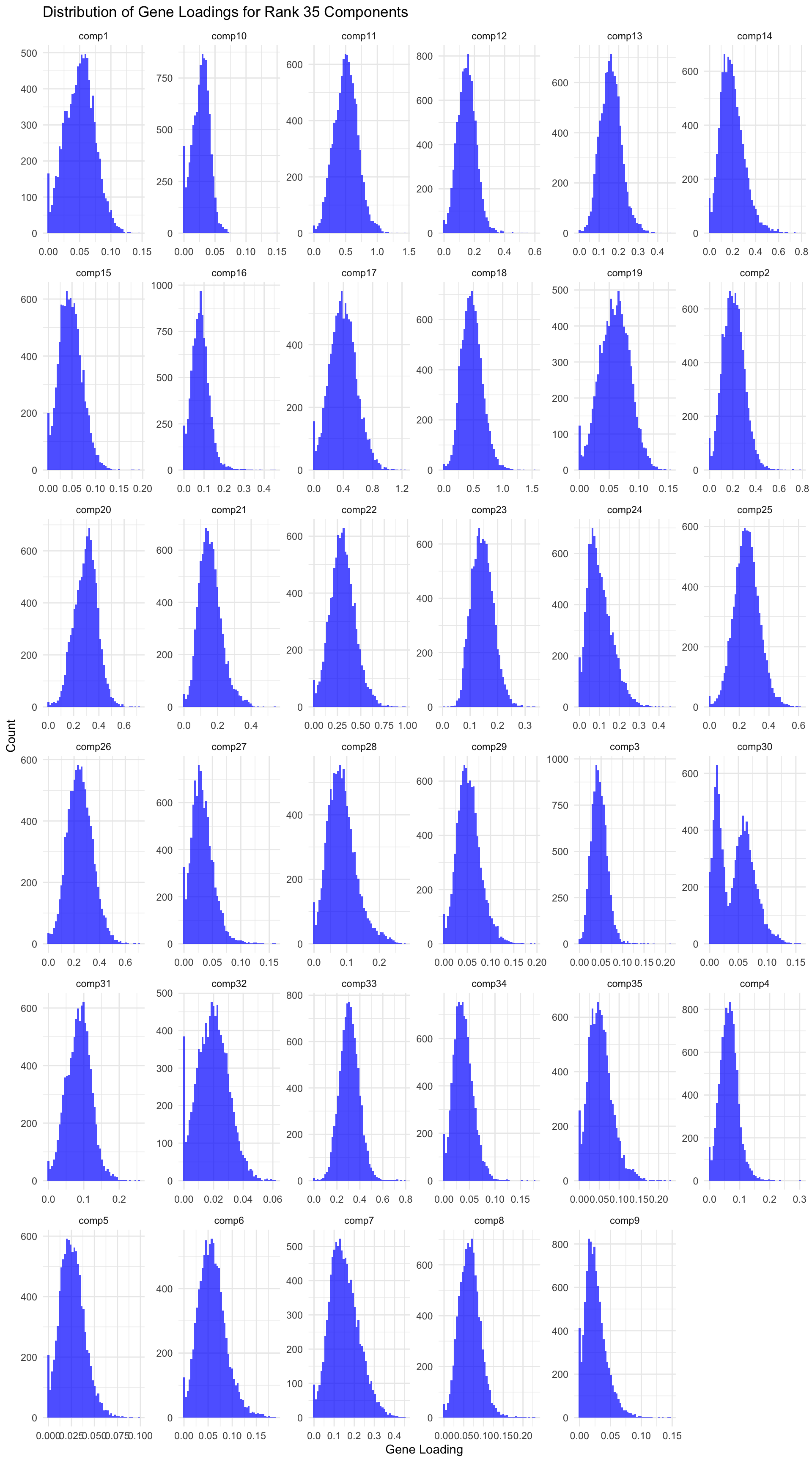
# Load data gene_factors <-read_csv("https://gannet.fish.washington.edu/v1_web/owlshell/bu-github/timeseries_molecular/M-multi-species/output/41-rank35-optimization/lambda_gene_1/barnacle_factors/gene_factors.csv")#edit so columns names are shifted 1 to the leftcolnames(gene_factors) <-c("gene", paste0("comp", 1:(ncol(gene_factors)-1)))#code to create histogram of gene_factors:library(ggplot2)library(tidyr)# Reshape data to long format gene_factors_long <- gene_factors %>%pivot_longer(cols =-1, names_to ="component", values_to ="loading")# Plot histogramggplot(gene_factors_long, aes(x = loading)) +geom_histogram(bins =50, fill ="blue", alpha =0.7) +facet_wrap(~ component, scales ="free") +theme_minimal() +labs(title ="Distribution of Gene Loadings for Rank 35 Components",x ="Gene Loading",y ="Count")
# Load data gene_factors <-read_csv("https://gannet.fish.washington.edu/v1_web/owlshell/bu-github/timeseries_molecular/M-multi-species/output/41-rank35-optimization/lambda_gene_2/barnacle_factors/gene_factors.csv")#edit so columns names are shifted 1 to the leftcolnames(gene_factors) <-c("gene", paste0("comp", 1:(ncol(gene_factors)-1)))#code to create histogram of gene_factors:library(ggplot2)library(tidyr)# Reshape data to long format gene_factors_long <- gene_factors %>%pivot_longer(cols =-1, names_to ="component", values_to ="loading")# Plot histogramggplot(gene_factors_long, aes(x = loading)) +geom_histogram(bins =50, fill ="blue", alpha =0.7) +facet_wrap(~ component, scales ="free") +theme_minimal() +labs(title ="Distribution of Gene Loadings for Rank 35 Components",x ="Gene Loading",y ="Count")
# Load data gene_factors <-read_csv("https://gannet.fish.washington.edu/v1_web/owlshell/bu-github/timeseries_molecular/M-multi-species/output/41-rank35-optimization/lambda_gene_4/barnacle_factors/gene_factors.csv")#edit so columns names are shifted 1 to the leftcolnames(gene_factors) <-c("gene", paste0("comp", 1:(ncol(gene_factors)-1)))#code to create histogram of gene_factors:library(ggplot2)library(tidyr)# Reshape data to long format gene_factors_long <- gene_factors %>%pivot_longer(cols =-1, names_to ="component", values_to ="loading")# Plot histogramggplot(gene_factors_long, aes(x = loading)) +geom_histogram(bins =50, fill ="blue", alpha =0.7) +facet_wrap(~ component, scales ="free") +theme_minimal() +labs(title ="Distribution of Gene Loadings for Rank 35 Components",x ="Gene Loading",y ="Count")
# Load data gene_factors <-read_csv("https://gannet.fish.washington.edu/v1_web/owlshell/bu-github/timeseries_molecular/M-multi-species/output/41-rank35-optimization/lambda_gene_6/barnacle_factors/gene_factors.csv")#edit so columns names are shifted 1 to the leftcolnames(gene_factors) <-c("gene", paste0("comp", 1:(ncol(gene_factors)-1)))#code to create histogram of gene_factors:library(ggplot2)library(tidyr)# Reshape data to long format gene_factors_long <- gene_factors %>%pivot_longer(cols =-1, names_to ="component", values_to ="loading")# Plot histogramggplot(gene_factors_long, aes(x = loading)) +geom_histogram(bins =50, fill ="blue", alpha =0.7) +facet_wrap(~ component, scales ="free") +theme_minimal() +labs(title ="Distribution of Gene Loadings for Rank 35 Components",x ="Gene Loading",y ="Count")
# Load data gene_factors <-read_csv("https://gannet.fish.washington.edu/v1_web/owlshell/bu-github/timeseries_molecular/M-multi-species/output/41-rank35-optimization/lambda_gene_8/barnacle_factors/gene_factors.csv")#edit so columns names are shifted 1 to the leftcolnames(gene_factors) <-c("gene", paste0("comp", 1:(ncol(gene_factors)-1)))#code to create histogram of gene_factors:library(ggplot2)library(tidyr)# Reshape data to long format gene_factors_long <- gene_factors %>%pivot_longer(cols =-1, names_to ="component", values_to ="loading")# Plot histogramggplot(gene_factors_long, aes(x = loading)) +geom_histogram(bins =50, fill ="blue", alpha =0.7) +facet_wrap(~ component, scales ="free") +theme_minimal() +labs(title ="Distribution of Gene Loadings for Rank 35 Components",x ="Gene Loading",y ="Count")
# Load data gene_factors <-read_csv("https://gannet.fish.washington.edu/v1_web/owlshell/bu-github/timeseries_molecular/M-multi-species/output/41-rank35-optimization/lambda_gene_10/barnacle_factors/gene_factors.csv")#edit so columns names are shifted 1 to the leftcolnames(gene_factors) <-c("gene", paste0("comp", 1:(ncol(gene_factors)-1)))#code to create histogram of gene_factors:library(ggplot2)library(tidyr)# Reshape data to long format gene_factors_long <- gene_factors %>%pivot_longer(cols =-1, names_to ="component", values_to ="loading")# Plot histogramggplot(gene_factors_long, aes(x = loading)) +geom_histogram(bins =50, fill ="blue", alpha =0.7) +facet_wrap(~ component, scales ="free") +theme_minimal() +labs(title ="Distribution of Gene Loadings for Rank 35 Components",x ="Gene Loading",y ="Count")
Source Code
---title: "Barnacle"description: "more lamda values"categories: ["Transcriptomics"]#citation: date: 12-04-2025image: http://gannet.fish.washington.edu/seashell/snaps/Monosnap_Image_2025-11-30_15-34-25.png # finding a good imageauthor: - name: Steven Roberts url: orcid: 0000-0001-8302-1138 affiliation: Professor, UW - School of Aquatic and Fishery Sciences affiliation-url: https://robertslab.info #url: # self-defineddraft: false # setting this to `true` will prevent your post from appearing on your listing page until you're ready!format: html: toc: true # ← enable TOC toc-location: left # or: right, body toc-depth: 3 # how many heading levels to show code-fold: FALSE code-tools: true code-copy: true highlight-style: github code-overflow: wrap#runtime: shiny---# More lamda testing``` bashLAMBDAS_GENE="1 2 4 6 8 10"LAMBDA_SAMPLE=0.1LAMBDA_TIME=0.05RANK=35mkdir-p ../output/41-rank35-optimizationOUTDIR_BASE=../output/41-rank35-optimizationfor LG in$LAMBDAS_GENE;doOUTDIR=${OUTDIR_BASE}/lambda_gene_${LG}mkdir-p"$OUTDIR"uv run python ../scripts/14.1-barnacle/build_tensor_and_run.py \--input-file ../output/14-pca-orthologs/vst_counts_matrix.csv \--output-dir"$OUTDIR"\--rank$RANK\--lambda-gene$LG\--lambda-sample$LAMBDA_SAMPLE\--lambda-time$LAMBDA_TIME\--max-iter 1000 \--tol 1e-5 \--seed 92done``````{r, echo=TRUE, warning=FALSE, message=FALSE}library(tidyverse)# ---------------------------------------------------------# SETTINGS# ---------------------------------------------------------base_url <- "https://gannet.fish.washington.edu/v1_web/owlshell/bu-github/timeseries_molecular/M-multi-species/output/41-rank35-optimization"lambda_vals <- c("1", "2", "4", "6", "8", "10")# ---------------------------------------------------------# FUNCTION: read gene_factors.csv AND FIX HEADER# ---------------------------------------------------------read_gene_factors <- function(lambda) { url <- sprintf("%s/lambda_gene_%s/barnacle_factors/gene_factors.csv", base_url, lambda) message("Reading: ", url) df <- read_csv(url, show_col_types = FALSE) # Fix the shifted header here # First column should be "gene", others "comp1..compN" colnames(df) <- c("gene", paste0("comp", 1:(ncol(df)-1))) df$lambda_gene <- lambda df}# ---------------------------------------------------------# READ ALL RUNS# ---------------------------------------------------------all_factors <- map_df(lambda_vals, read_gene_factors)# ---------------------------------------------------------# LONG FORMAT FOR ANALYSIS# ---------------------------------------------------------factor_long <- all_factors %>% pivot_longer( cols = starts_with("comp"), names_to = "component", values_to = "loading" )``````{r}sparsity_tbl <- factor_long %>%group_by(lambda_gene) %>%summarize(n_values =n(),n_zero =sum(loading ==0|abs(loading) <1e-12),sparsity = n_zero / n_values )sparsity_tbl ``````{r}gene_component_counts <- factor_long %>%filter(loading >0) %>%group_by(lambda_gene, gene) %>%summarize(n_components =n_distinct(component),.groups ="drop" )``````{r}multi_component_summary <- gene_component_counts %>%group_by(lambda_gene, n_components) %>%summarize(n_genes =n(), .groups ="drop")multi_component_summary``````{r}summary_tbl <- sparsity_tbl %>%left_join( gene_component_counts %>%group_by(lambda_gene) %>%summarize(mean_components =mean(n_components),frac_multi =mean(n_components >1),.groups ="drop" ),by ="lambda_gene" )summary_tbl``````{r, echo=TRUE, warning=FALSE, message=FALSE, fig.width=10, fig.height=18} # Load data gene_factors <- read_csv("https://gannet.fish.washington.edu/v1_web/owlshell/bu-github/timeseries_molecular/M-multi-species/output/41-rank35-optimization/lambda_gene_1/barnacle_factors/gene_factors.csv")#edit so columns names are shifted 1 to the left colnames(gene_factors) <- c("gene", paste0("comp", 1:(ncol(gene_factors)-1)))#code to create histogram of gene_factors: library(ggplot2) library(tidyr) # Reshape data to long format gene_factors_long <- gene_factors %>% pivot_longer(cols = -1, names_to = "component", values_to = "loading") # Plot histogram ggplot(gene_factors_long, aes(x = loading)) + geom_histogram(bins = 50, fill = "blue", alpha = 0.7) + facet_wrap(~ component, scales = "free") + theme_minimal() + labs(title = "Distribution of Gene Loadings for Rank 35 Components", x = "Gene Loading", y = "Count")``````{r, echo=TRUE, warning=FALSE, message=FALSE, fig.width=10, fig.height=18} # Load data gene_factors <- read_csv("https://gannet.fish.washington.edu/v1_web/owlshell/bu-github/timeseries_molecular/M-multi-species/output/41-rank35-optimization/lambda_gene_2/barnacle_factors/gene_factors.csv")#edit so columns names are shifted 1 to the left colnames(gene_factors) <- c("gene", paste0("comp", 1:(ncol(gene_factors)-1)))#code to create histogram of gene_factors: library(ggplot2) library(tidyr) # Reshape data to long format gene_factors_long <- gene_factors %>% pivot_longer(cols = -1, names_to = "component", values_to = "loading") # Plot histogram ggplot(gene_factors_long, aes(x = loading)) + geom_histogram(bins = 50, fill = "blue", alpha = 0.7) + facet_wrap(~ component, scales = "free") + theme_minimal() + labs(title = "Distribution of Gene Loadings for Rank 35 Components", x = "Gene Loading", y = "Count")``````{r, echo=TRUE, warning=FALSE, message=FALSE, fig.width=10, fig.height=18} # Load data gene_factors <- read_csv("https://gannet.fish.washington.edu/v1_web/owlshell/bu-github/timeseries_molecular/M-multi-species/output/41-rank35-optimization/lambda_gene_4/barnacle_factors/gene_factors.csv")#edit so columns names are shifted 1 to the left colnames(gene_factors) <- c("gene", paste0("comp", 1:(ncol(gene_factors)-1)))#code to create histogram of gene_factors: library(ggplot2) library(tidyr) # Reshape data to long format gene_factors_long <- gene_factors %>% pivot_longer(cols = -1, names_to = "component", values_to = "loading") # Plot histogram ggplot(gene_factors_long, aes(x = loading)) + geom_histogram(bins = 50, fill = "blue", alpha = 0.7) + facet_wrap(~ component, scales = "free") + theme_minimal() + labs(title = "Distribution of Gene Loadings for Rank 35 Components", x = "Gene Loading", y = "Count")``````{r, echo=TRUE, warning=FALSE, message=FALSE, fig.width=10, fig.height=18} # Load data gene_factors <- read_csv("https://gannet.fish.washington.edu/v1_web/owlshell/bu-github/timeseries_molecular/M-multi-species/output/41-rank35-optimization/lambda_gene_6/barnacle_factors/gene_factors.csv")#edit so columns names are shifted 1 to the left colnames(gene_factors) <- c("gene", paste0("comp", 1:(ncol(gene_factors)-1)))#code to create histogram of gene_factors: library(ggplot2) library(tidyr) # Reshape data to long format gene_factors_long <- gene_factors %>% pivot_longer(cols = -1, names_to = "component", values_to = "loading") # Plot histogram ggplot(gene_factors_long, aes(x = loading)) + geom_histogram(bins = 50, fill = "blue", alpha = 0.7) + facet_wrap(~ component, scales = "free") + theme_minimal() + labs(title = "Distribution of Gene Loadings for Rank 35 Components", x = "Gene Loading", y = "Count")``````{r, echo=TRUE, warning=FALSE, message=FALSE, fig.width=10, fig.height=18} # Load data gene_factors <- read_csv("https://gannet.fish.washington.edu/v1_web/owlshell/bu-github/timeseries_molecular/M-multi-species/output/41-rank35-optimization/lambda_gene_8/barnacle_factors/gene_factors.csv")#edit so columns names are shifted 1 to the left colnames(gene_factors) <- c("gene", paste0("comp", 1:(ncol(gene_factors)-1)))#code to create histogram of gene_factors: library(ggplot2) library(tidyr) # Reshape data to long format gene_factors_long <- gene_factors %>% pivot_longer(cols = -1, names_to = "component", values_to = "loading") # Plot histogram ggplot(gene_factors_long, aes(x = loading)) + geom_histogram(bins = 50, fill = "blue", alpha = 0.7) + facet_wrap(~ component, scales = "free") + theme_minimal() + labs(title = "Distribution of Gene Loadings for Rank 35 Components", x = "Gene Loading", y = "Count")``````{r, echo=TRUE, warning=FALSE, message=FALSE, fig.width=10, fig.height=18} # Load data gene_factors <- read_csv("https://gannet.fish.washington.edu/v1_web/owlshell/bu-github/timeseries_molecular/M-multi-species/output/41-rank35-optimization/lambda_gene_10/barnacle_factors/gene_factors.csv")#edit so columns names are shifted 1 to the left colnames(gene_factors) <- c("gene", paste0("comp", 1:(ncol(gene_factors)-1)))#code to create histogram of gene_factors: library(ggplot2) library(tidyr) # Reshape data to long format gene_factors_long <- gene_factors %>% pivot_longer(cols = -1, names_to = "component", values_to = "loading") # Plot histogram ggplot(gene_factors_long, aes(x = loading)) + geom_histogram(bins = 50, fill = "blue", alpha = 0.7) + facet_wrap(~ component, scales = "free") + theme_minimal() + labs(title = "Distribution of Gene Loadings for Rank 35 Components", x = "Gene Loading", y = "Count")```