
10-Hisat
Steven Roberts 22 May, 2024
Alt splice test run
1 Differentially Expressed Transcripts
2 Reads
ls ../data/reads/*../data/reads/100.trimmed.R1.fastq.gz
../data/reads/100.trimmed.R2.fastq.gz
../data/reads/107.trimmed.R1.fastq.gz
../data/reads/107.trimmed.R2.fastq.gz
../data/reads/108.trimmed.R1.fastq.gz
../data/reads/108.trimmed.R2.fastq.gz
../data/reads/109.trimmed.R1.fastq.gz
../data/reads/109.trimmed.R2.fastq.gz
../data/reads/10.trimmed.R1.fastq.gz
../data/reads/10.trimmed.R2.fastq.gz
../data/reads/110.trimmed.R1.fastq.gz
../data/reads/110.trimmed.R2.fastq.gz
../data/reads/116.trimmed.R1.fastq.gz
../data/reads/116.trimmed.R2.fastq.gz
../data/reads/11.trimmed.R1.fastq.gz
../data/reads/11.trimmed.R2.fastq.gz
../data/reads/12.trimmed.R1.fastq.gz
../data/reads/12.trimmed.R2.fastq.gz
../data/reads/13.trimmed.R1.fastq.gz
../data/reads/13.trimmed.R2.fastq.gz
../data/reads/18.trimmed.R1.fastq.gz
../data/reads/18.trimmed.R2.fastq.gz
../data/reads/19.trimmed.R1.fastq.gz
../data/reads/19.trimmed.R2.fastq.gz
../data/reads/1.trimmed.R1.fastq.gz
../data/reads/1.trimmed.R2.fastq.gz
../data/reads/20.trimmed.R1.fastq.gz
../data/reads/20.trimmed.R2.fastq.gz
../data/reads/21.trimmed.R1.fastq.gz
../data/reads/21.trimmed.R2.fastq.gz
../data/reads/28.trimmed.R1.fastq.gz
../data/reads/28.trimmed.R2.fastq.gz
../data/reads/29.trimmed.R1.fastq.gz
../data/reads/29.trimmed.R2.fastq.gz
../data/reads/2.trimmed.R1.fastq.gz
../data/reads/2.trimmed.R2.fastq.gz
../data/reads/30.trimmed.R1.fastq.gz
../data/reads/30.trimmed.R2.fastq.gz
../data/reads/31.trimmed.R1.fastq.gz
../data/reads/31.trimmed.R2.fastq.gz
../data/reads/36.trimmed.R1.fastq.gz
../data/reads/36.trimmed.R2.fastq.gz
../data/reads/3.trimmed.R1.fastq.gz
../data/reads/3.trimmed.R2.fastq.gz
../data/reads/4.trimmed.R1.fastq.gz
../data/reads/4.trimmed.R2.fastq.gz
../data/reads/5.trimmed.R1.fastq.gz
../data/reads/5.trimmed.R2.fastq.gz
../data/reads/78.trimmed.R1.fastq.gz
../data/reads/78.trimmed.R2.fastq.gz
../data/reads/79.trimmed.R1.fastq.gz
../data/reads/79.trimmed.R2.fastq.gz
../data/reads/80.trimmed.R1.fastq.gz
../data/reads/80.trimmed.R2.fastq.gz
../data/reads/83.trimmed.R1.fastq.gz
../data/reads/83.trimmed.R2.fastq.gz
../data/reads/88.trimmed.R1.fastq.gz
../data/reads/88.trimmed.R2.fastq.gz
../data/reads/90.trimmed.R1.fastq.gz
../data/reads/90.trimmed.R2.fastq.gz
../data/reads/91.trimmed.R1.fastq.gz
../data/reads/91.trimmed.R2.fastq.gz
../data/reads/92.trimmed.R1.fastq.gz
../data/reads/92.trimmed.R2.fastq.gz
../data/reads/94.trimmed.R1.fastq.gz
../data/reads/94.trimmed.R2.fastq.gz
../data/reads/97.trimmed.R1.fastq.gz
../data/reads/97.trimmed.R2.fastq.gz
../data/reads/98.trimmed.R1.fastq.gz
../data/reads/98.trimmed.R2.fastq.gz
../data/reads/99.trimmed.R1.fastq.gz
../data/reads/99.trimmed.R2.fastq.gz
../data/reads/splice-test-files.txt3 Genome
https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_031168955.1/
cd ../data
/home/shared/datasets download genome accession GCF_031168955.1 --include gff3,gtf,rna,cds,protein,genome,seq-reportcd ../data
unzip ncbi_dataset.zipls ../data/ncbi_dataset/data/GCF_031168955.1cds_from_genomic.fna
GCF_031168955.1_ASM3116895v1_genomic.fna
genomic.gff
genomic.gtf
protein.faa
rna.fna
sequence_report.jsonl4 Hisat
/home/shared/hisat2-2.2.1/hisat2_extract_exons.py \
../data/ncbi_dataset/data/GCF_031168955.1/genomic.gtf \
> ../output/10.1-hisat-deseq2/m_exon.tab/home/shared/hisat2-2.2.1/hisat2_extract_splice_sites.py \
../data/ncbi_dataset/data/GCF_031168955.1/genomic.gtf \
> ../output/10.1-hisat-deseq2/m_spice_sites.tabWill go ahead and exclude index from git
echo "10.1-hisat-deseq2/GCF*" >> ../output/.gitignore/home/shared/hisat2-2.2.1/hisat2-build \
../data/ncbi_dataset/data/GCF_031168955.1/GCF_031168955.1_ASM3116895v1_genomic.fna \
../output/10.1-hisat-deseq2/GCF_031168955.1.index \
--exon ../output/10.1-hisat-deseq2/m_exon.tab \
--ss ../output/10.1-hisat-deseq2/m_spice_sites.tab \
-p 36 \
../data/ncbi_dataset/data/GCF_031168955.1/genomic.gtf \
2> ../output/10.1-hisat-deseq2/hisat2-build_stats.txtecho "10.1-hisat-deseq2/*sam" >> ../output/.gitignorefind ../data/reads/*.trimmed.R1.fastq.gz \
| xargs basename -s .trimmed.R1.fastq.gz | xargs -I{} \
/home/shared/hisat2-2.2.1/hisat2 \
-x ../output/10.1-hisat-deseq2/GCF_031168955.1.index \
--dta \
-p 20 \
-1 ../data/reads/{}.trimmed.R1.fastq.gz \
-2 ../data/reads/{}.trimmed.R2.fastq.gz \
-S ../output/10.1-hisat-deseq2/{}.sam \
2> ../output/10.1-hisat-deseq2/hisat.outecho "10.1-hisat-deseq2/*bam" >> ../output/.gitignore
echo "10.1-hisat-deseq2/*bam*" >> ../output/.gitignorefor samfile in ../output/10.1-hisat-deseq2/*.sam; do
bamfile="${samfile%.sam}.bam"
sorted_bamfile="${samfile%.sam}.sorted.bam"
/home/shared/samtools-1.12/samtools view -bS -@ 20 "$samfile" > "$bamfile"
/home/shared/samtools-1.12/samtools sort -@ 20 "$bamfile" -o "$sorted_bamfile"
/home/shared/samtools-1.12/samtools index -@ 20 "$sorted_bamfile"
donerm ../output/10.1-hisat-deseq2/*samls ../output/10.1-hisat-deseq2/*sorted.bam | wc -ltail ../output/10.1-hisat-deseq2/hisat.out ----
3794910 pairs aligned concordantly 0 times; of these:
146293 (3.85%) aligned discordantly 1 time
----
3648617 pairs aligned 0 times concordantly or discordantly; of these:
7297234 mates make up the pairs; of these:
5640288 (77.29%) aligned 0 times
1498025 (20.53%) aligned exactly 1 time
158921 (2.18%) aligned >1 times
82.81% overall alignment ratecat ../output/10.1-hisat-deseq2/hisat.out \
| grep "overall alignment rate"87.53% overall alignment rate
86.32% overall alignment rate
81.37% overall alignment rate
86.39% overall alignment rate
83.54% overall alignment rate
87.67% overall alignment rate
86.69% overall alignment rate
86.56% overall alignment rate
84.58% overall alignment rate
81.26% overall alignment rate
83.03% overall alignment rate
86.29% overall alignment rate
84.82% overall alignment rate
82.29% overall alignment rate
83.16% overall alignment rate
86.14% overall alignment rate
81.04% overall alignment rate
87.28% overall alignment rate
84.89% overall alignment rate
87.76% overall alignment rate
88.45% overall alignment rate
83.13% overall alignment rate
83.21% overall alignment rate
84.99% overall alignment rate
86.33% overall alignment rate
86.47% overall alignment rate
84.37% overall alignment rate
87.60% overall alignment rate
75.31% overall alignment rate
81.73% overall alignment rate
84.47% overall alignment rate
80.84% overall alignment rate
86.72% overall alignment rate
87.60% overall alignment rate
81.89% overall alignment rate
82.81% overall alignment rate5 Stringtie
echo "10.1-hisat-deseq2/*gtf" >> ../output/.gitignorefind ../output/10.1-hisat-deseq2/*sorted.bam \
| xargs basename -s .sorted.bam | xargs -I{} \
/home/shared/stringtie-2.2.1.Linux_x86_64/stringtie \
-p 36 \
-eB \
-G ../data/ncbi_dataset/data/GCF_031168955.1/genomic.gff \
-o ../output/10.1-hisat-deseq2/{}.gtf \
../output/10.1-hisat-deseq2/{}.sorted.bam6 Count Matrix
ls ../output/10.1-hisat-deseq2/*gtf../output/10.1-hisat-deseq2/100.gtf
../output/10.1-hisat-deseq2/107.gtf
../output/10.1-hisat-deseq2/108.gtf
../output/10.1-hisat-deseq2/109.gtf
../output/10.1-hisat-deseq2/10.gtf
../output/10.1-hisat-deseq2/110.gtf
../output/10.1-hisat-deseq2/116.gtf
../output/10.1-hisat-deseq2/11.gtf
../output/10.1-hisat-deseq2/12.gtf
../output/10.1-hisat-deseq2/13.gtf
../output/10.1-hisat-deseq2/18.gtf
../output/10.1-hisat-deseq2/19.gtf
../output/10.1-hisat-deseq2/1.gtf
../output/10.1-hisat-deseq2/20.gtf
../output/10.1-hisat-deseq2/21.gtf
../output/10.1-hisat-deseq2/28.gtf
../output/10.1-hisat-deseq2/29.gtf
../output/10.1-hisat-deseq2/2.gtf
../output/10.1-hisat-deseq2/30.gtf
../output/10.1-hisat-deseq2/31.gtf
../output/10.1-hisat-deseq2/36.gtf
../output/10.1-hisat-deseq2/3.gtf
../output/10.1-hisat-deseq2/4.gtf
../output/10.1-hisat-deseq2/5.gtf
../output/10.1-hisat-deseq2/78.gtf
../output/10.1-hisat-deseq2/79.gtf
../output/10.1-hisat-deseq2/80.gtf
../output/10.1-hisat-deseq2/83.gtf
../output/10.1-hisat-deseq2/88.gtf
../output/10.1-hisat-deseq2/90.gtf
../output/10.1-hisat-deseq2/91.gtf
../output/10.1-hisat-deseq2/92.gtf
../output/10.1-hisat-deseq2/94.gtf
../output/10.1-hisat-deseq2/97.gtf
../output/10.1-hisat-deseq2/98.gtf
../output/10.1-hisat-deseq2/99.gtfcat ../output/10.1-hisat-deseq2/list01.txt1 ../output/10-hisat-deseq2/1.gtf
2 ../output/10-hisat-deseq2/2.gtf
3 ../output/10-hisat-deseq2/3.gtf
4 ../output/10-hisat-deseq2/4.gtf
5 ../output/10-hisat-deseq2/5.gtf
10 ../output/10-hisat-deseq2/10.gtf
11 ../output/10-hisat-deseq2/11.gtf
12 ../output/10-hisat-deseq2/12.gtf
13 ../output/10-hisat-deseq2/13.gtf
18 ../output/10-hisat-deseq2/18.gtf
19 ../output/10-hisat-deseq2/19.gtf
20 ../output/10-hisat-deseq2/20.gtf
21 ../output/10-hisat-deseq2/21.gtf
28 ../output/10-hisat-deseq2/28.gtf
29 ../output/10-hisat-deseq2/29.gtf
30 ../output/10-hisat-deseq2/30.gtf
31 ../output/10-hisat-deseq2/31.gtf
36 ../output/10-hisat-deseq2/36.gtf
78 ../output/10-hisat-deseq2/78.gtf
79 ../output/10-hisat-deseq2/79.gtf
80 ../output/10-hisat-deseq2/80.gtf
83 ../output/10-hisat-deseq2/83.gtf
88 ../output/10-hisat-deseq2/88.gtf
90 ../output/10-hisat-deseq2/90.gtf
91 ../output/10-hisat-deseq2/91.gtf
92 ../output/10-hisat-deseq2/92.gtf
94 ../output/10-hisat-deseq2/94.gtf
97 ../output/10-hisat-deseq2/97.gtf
98 ../output/10-hisat-deseq2/98.gtf
99 ../output/10-hisat-deseq2/99.gtf
100 ../output/10-hisat-deseq2/100.gtf
107 ../output/10-hisat-deseq2/107.gtf
108 ../output/10-hisat-deseq2/108.gtf
109 ../output/10-hisat-deseq2/109.gtf
110 ../output/10-hisat-deseq2/110.gtf
116 ../output/10-hisat-deseq2/116.gtfpython /home/shared/stringtie-2.2.1.Linux_x86_64/prepDE.py \
-i ../output/10.1-hisat-deseq2/list01.txt \
-g ../output/10.1-hisat-deseq2/gene_count_matrix.csv \
-t ../output/10.1-hisat-deseq2/transcript_count_matrix.csvhead ../output/10.1-hisat-deseq2/*matrix.csv==> ../output/10.1-hisat-deseq2/gene_count_matrix.csv <==
gene_id,1,10,100,107,108,109,11,110,116,12,13,18,19,2,20,21,28,29,3,30,31,36,4,5,78,79,80,83,88,90,91,92,94,97,98,99
gene-LOC132462341|LOC132462341,360,464,391,346,691,408,436,509,366,373,432,385,330,288,307,346,293,347,452,452,984,230,469,400,472,311,368,312,551,631,605,366,339,517,577,235
gene-abce1|abce1,694,325,276,77,409,196,281,284,327,310,393,386,106,290,252,326,345,254,363,353,922,160,588,285,178,119,197,134,128,103,315,204,301,56,49,260
gene-si:dkey-6i22.5|si:dkey-6i22.5,0,10,13,11,49,42,14,36,0,14,9,0,0,14,3,21,26,9,39,20,10,0,0,10,48,41,60,39,49,54,20,27,7,22,63,12
gene-ube2v1|ube2v1,10,22,15,19,74,28,63,35,30,31,25,42,17,14,21,60,61,14,29,37,68,37,0,30,10,13,56,32,45,55,27,38,60,25,18,50
gene-cldn15la|cldn15la,0,9,0,0,18,0,0,0,0,27,0,0,0,0,0,0,0,0,4,35,0,0,0,4,0,0,0,0,8,32,0,0,14,0,0,0
gene-muc15|muc15,0,40,40,29,8,0,38,0,16,15,0,0,0,0,22,22,69,22,0,15,0,34,55,51,0,30,19,31,0,0,42,34,0,0,26,56
gene-pcloa|pcloa,0,3,0,0,41,7,4,0,3,0,0,0,27,0,0,0,0,4,0,10,0,23,0,0,0,0,4,4,0,0,4,0,0,22,101,0
gene-LOC132472829|LOC132472829,402,550,293,182,1082,213,234,149,444,360,531,242,269,194,359,383,395,392,408,489,310,352,168,403,462,287,310,113,457,321,396,234,455,279,229,208
gene-ifi35|ifi35,3696,2492,2615,2184,2702,2579,3631,3158,2121,2673,2893,2210,2324,2384,2561,2654,2887,2250,3143,2830,1297,1981,2971,2651,2669,2671,1790,2173,2603,3260,2186,2451,2464,2768,1935,1991
==> ../output/10.1-hisat-deseq2/transcript_count_matrix.csv <==
transcript_id,1,10,100,107,108,109,11,110,116,12,13,18,19,2,20,21,28,29,3,30,31,36,4,5,78,79,80,83,88,90,91,92,94,97,98,99
rna-XM_060037252.1,20,918,84,11,385,70,54,82,131,72,52,8,26,41,33,62,30,12,45,85,377,59,58,57,96,19,55,94,92,125,54,34,69,36,16,83
rna-XM_060070002.1,0,0,0,0,0,12,0,0,0,14,14,0,0,0,0,0,0,3,0,0,0,0,0,0,0,0,0,0,0,0,13,0,0,0,0,7
rna-XM_060056394.1,0,0,0,1,0,0,336,0,37,0,0,0,86,0,0,0,0,0,395,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,1
rna-XM_060072836.1,0,0,14,4,0,0,5,0,15,0,0,0,4,0,0,5,0,0,0,11,0,0,5,0,0,12,0,0,12,3,10,0,9,0,0,0
rna-XM_060062883.1,719,38,380,279,652,462,461,381,432,0,831,479,641,171,593,82,520,625,366,533,640,311,36,876,365,322,351,342,430,324,450,410,540,336,513,370
rna-XM_060057326.1,1072,1577,801,1056,1420,909,1523,1101,951,1459,2162,1170,1983,1317,1264,1398,2075,1055,1303,1820,1496,934,966,1509,1132,928,845,948,1930,2047,1039,781,979,2259,2747,740
rna-XM_060051796.1,0,1,1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,3,0,0,0,0,0,0,0,4,0,2,0,0,0,0,0,0,0,2
rna-XM_060076321.1,0,0,0,0,16,0,0,0,0,0,0,4,58,0,0,0,0,0,0,12,0,0,0,0,0,0,0,0,0,0,0,0,5,13,80,0
rna-XM_060064475.1,6,1771,834,10,33,0,1752,997,470,1572,0,0,16,713,0,0,0,5,0,679,292,656,0,1051,6,1326,0,0,0,0,592,448,0,0,0,57 DEseq2
cat ../output/10.1-hisat-deseq2/conditions.txtSampleID Condition
X1 16
X2 16
X3 16
X4 16
X5 16
X10 16
X11 16
X12 16
X13 16
X18 16
X19 16
X20 16
X21 16
X28 16
X29 16
X30 16
X31 16
X36 16
X78 9
X79 9
X80 9
X83 9
X88 9
X90 9
X91 9
X92 9
X94 9
X97 9
X98 9
X99 9
X100 9
X107 9
X108 9
X109 9
X110 9
X116 9library(DESeq2)# Load transcript) count matrix and labels
countData <- as.matrix(read.csv("../output/10.1-hisat-deseq2/transcript_count_matrix.csv", row.names="transcript_id"))
colData <- read.csv("../output/10.1-hisat-deseq2/conditions.txt", sep="\t", row.names = 1)
# Note: The PHENO_DATA file contains information on each sample, e.g., sex or population.
# The exact way to import this depends on the format of the file.
# Check all sample IDs in colData are also in CountData and match their orders
all(rownames(colData) %in% colnames(countData)) # This should return TRUE[1] TRUEcountData <- countData[, rownames(colData)]
all(rownames(colData) == colnames(countData)) # This should also return TRUE[1] TRUE# Create a DESeqDataSet from count matrix and labels
dds <- DESeqDataSetFromMatrix(countData = countData,
colData = colData, design = ~ Condition)
# Run the default analysis for DESeq2 and generate results table
dds <- DESeq(dds)
deseq2.res <- results(dds)
# Sort by adjusted p-value and display
resOrdered <- deseq2.res[order(deseq2.res$padj), ]
vsd <- vst(dds, blind = FALSE)
plotPCA(vsd, intgroup = "Condition")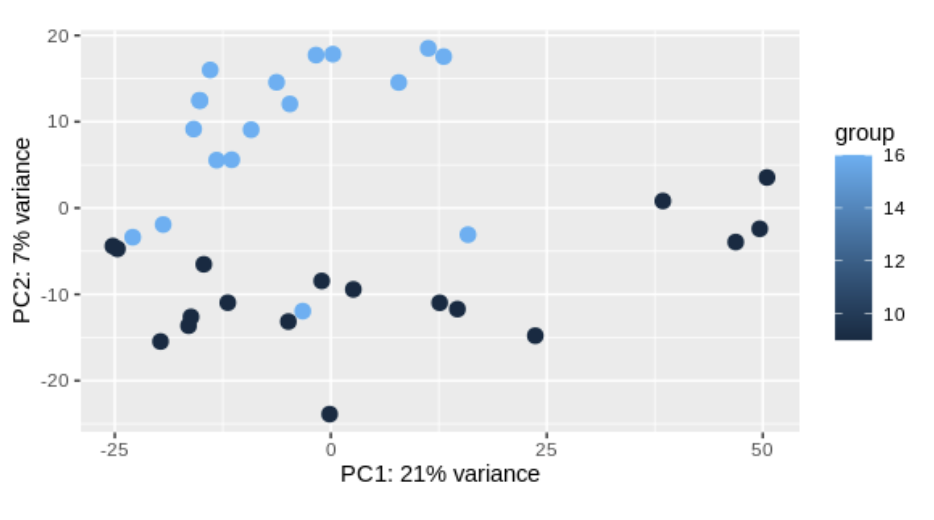
# Select top 50 differentially expressed genes
res <- results(dds)
res_ordered <- res[order(res$padj), ]
top_genes <- row.names(res_ordered)[1:50]
# Extract counts and normalize
counts <- counts(dds, normalized = TRUE)
counts_top <- counts[top_genes, ]
# Log-transform counts
log_counts_top <- log2(counts_top + 1)
# Generate heatmap
pheatmap(log_counts_top, scale = "row")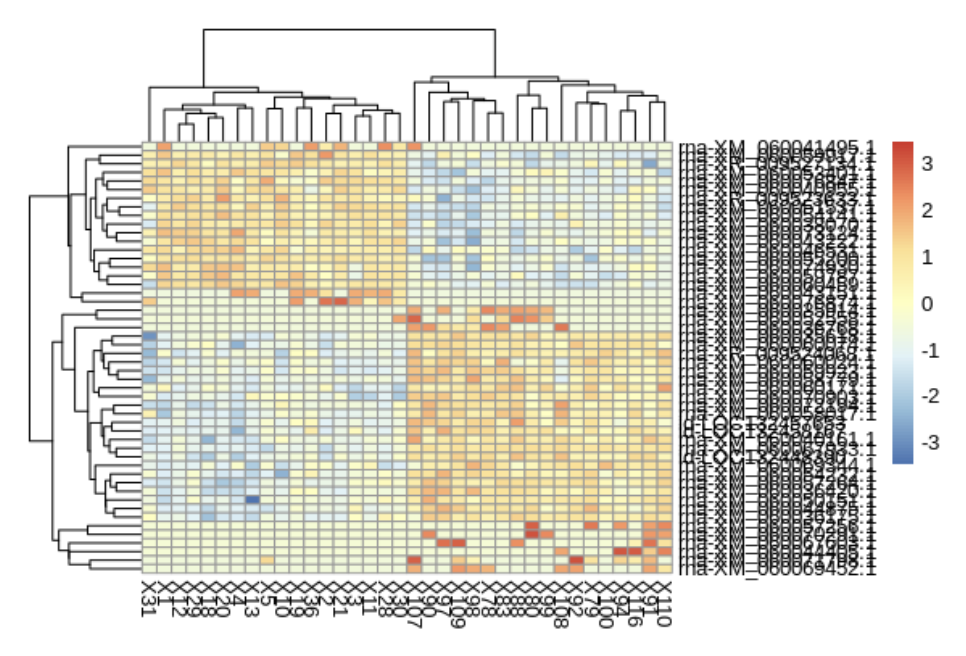
# Count number of hits with adjusted p-value less then 0.05
dim(res[!is.na(deseq2.res$padj) & deseq2.res$padj <= 0.05, ])[1] 4098 6tmp <- deseq2.res
# The main plot
plot(tmp$baseMean, tmp$log2FoldChange, pch=20, cex=0.45, ylim=c(-3, 3), log="x", col="darkgray",
main="DEG Dessication (pval <= 0.05)",
xlab="mean of normalized counts",
ylab="Log2 Fold Change")
# Getting the significant points and plotting them again so they're a different color
tmp.sig <- deseq2.res[!is.na(deseq2.res$padj) & deseq2.res$padj <= 0.05, ]
points(tmp.sig$baseMean, tmp.sig$log2FoldChange, pch=20, cex=0.45, col="red")
# 2 FC lines
abline(h=c(-1,1), col="blue")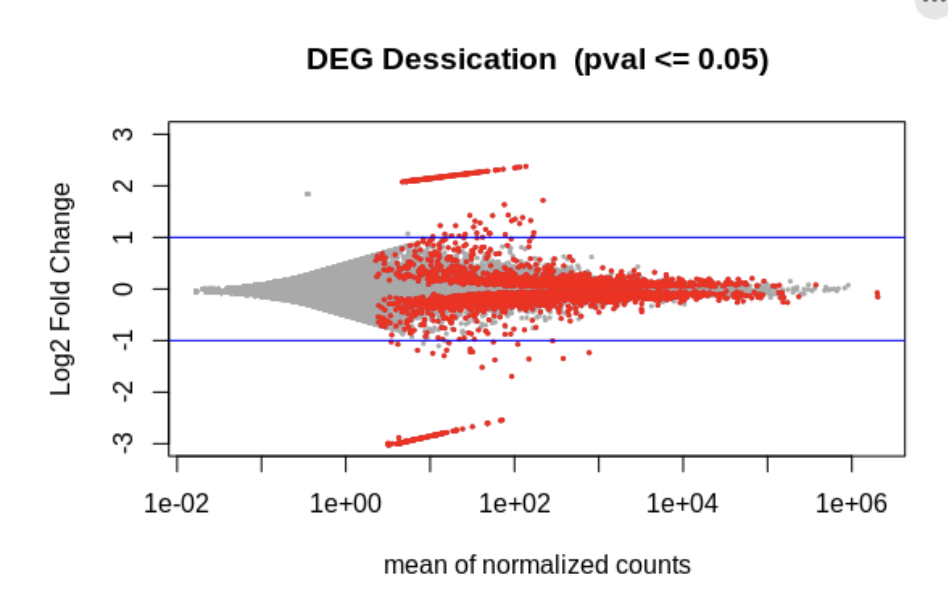
write.table(tmp.sig, "../output/10.1-hisat-deseq2/DETlist.tab", sep = '\t', row.names = T)detlist <- read.csv("../output/10.1-hisat-deseq2/DETlist.tab", sep = '\t', header = TRUE)
detlist$RowName <- rownames(detlist)
detlist2 <- detlist[, c("RowName", "pvalue")] # Optionally, reorder the columnshead(detlist) baseMean log2FoldChange lfcSE stat pvalue
rna-XM_060040999.1 327.23637 0.07503113 0.02706549 2.772206 5.567773e-03
rna-XM_060059475.1 64.57272 -0.09356216 0.03262081 -2.868174 4.128491e-03
rna-XM_060060617.1 42.88642 -0.23727065 0.04910582 -4.831823 1.352884e-06
rna-XM_060067174.1 237.63970 0.07702393 0.02363060 3.259500 1.116087e-03
rna-XM_060057172.1 37.57466 -0.20032720 0.06166216 -3.248787 1.158984e-03
rna-XM_060038089.1 703.71042 0.08841799 0.02025754 4.364695 1.273003e-05
padj RowName
rna-XM_060040999.1 4.170294e-02 rna-XM_060040999.1
rna-XM_060059475.1 3.329193e-02 rna-XM_060059475.1
rna-XM_060060617.1 4.605113e-05 rna-XM_060060617.1
rna-XM_060067174.1 1.243125e-02 rna-XM_060067174.1
rna-XM_060057172.1 1.279752e-02 rna-XM_060057172.1
rna-XM_060038089.1 3.171650e-04 rna-XM_060038089.1```