
1 Differentially Expressed Genes
Alt splice test run
2 Reads
ls ../data/reads/*../data/reads/100.trimmed.R1.fastq.gz
../data/reads/100.trimmed.R2.fastq.gz
../data/reads/107.trimmed.R1.fastq.gz
../data/reads/107.trimmed.R2.fastq.gz
../data/reads/108.trimmed.R1.fastq.gz
../data/reads/108.trimmed.R2.fastq.gz
../data/reads/109.trimmed.R1.fastq.gz
../data/reads/109.trimmed.R2.fastq.gz
../data/reads/10.trimmed.R1.fastq.gz
../data/reads/10.trimmed.R2.fastq.gz
../data/reads/110.trimmed.R1.fastq.gz
../data/reads/110.trimmed.R2.fastq.gz
../data/reads/116.trimmed.R1.fastq.gz
../data/reads/116.trimmed.R2.fastq.gz
../data/reads/11.trimmed.R1.fastq.gz
../data/reads/11.trimmed.R2.fastq.gz
../data/reads/12.trimmed.R1.fastq.gz
../data/reads/12.trimmed.R2.fastq.gz
../data/reads/13.trimmed.R1.fastq.gz
../data/reads/13.trimmed.R2.fastq.gz
../data/reads/18.trimmed.R1.fastq.gz
../data/reads/18.trimmed.R2.fastq.gz
../data/reads/19.trimmed.R1.fastq.gz
../data/reads/19.trimmed.R2.fastq.gz
../data/reads/1.trimmed.R1.fastq.gz
../data/reads/1.trimmed.R2.fastq.gz
../data/reads/20.trimmed.R1.fastq.gz
../data/reads/20.trimmed.R2.fastq.gz
../data/reads/21.trimmed.R1.fastq.gz
../data/reads/21.trimmed.R2.fastq.gz
../data/reads/28.trimmed.R1.fastq.gz
../data/reads/28.trimmed.R2.fastq.gz
../data/reads/29.trimmed.R1.fastq.gz
../data/reads/29.trimmed.R2.fastq.gz
../data/reads/2.trimmed.R1.fastq.gz
../data/reads/2.trimmed.R2.fastq.gz
../data/reads/30.trimmed.R1.fastq.gz
../data/reads/30.trimmed.R2.fastq.gz
../data/reads/31.trimmed.R1.fastq.gz
../data/reads/31.trimmed.R2.fastq.gz
../data/reads/36.trimmed.R1.fastq.gz
../data/reads/36.trimmed.R2.fastq.gz
../data/reads/3.trimmed.R1.fastq.gz
../data/reads/3.trimmed.R2.fastq.gz
../data/reads/4.trimmed.R1.fastq.gz
../data/reads/4.trimmed.R2.fastq.gz
../data/reads/5.trimmed.R1.fastq.gz
../data/reads/5.trimmed.R2.fastq.gz
../data/reads/78.trimmed.R1.fastq.gz
../data/reads/78.trimmed.R2.fastq.gz
../data/reads/79.trimmed.R1.fastq.gz
../data/reads/79.trimmed.R2.fastq.gz
../data/reads/80.trimmed.R1.fastq.gz
../data/reads/80.trimmed.R2.fastq.gz
../data/reads/83.trimmed.R1.fastq.gz
../data/reads/83.trimmed.R2.fastq.gz
../data/reads/88.trimmed.R1.fastq.gz
../data/reads/88.trimmed.R2.fastq.gz
../data/reads/90.trimmed.R1.fastq.gz
../data/reads/90.trimmed.R2.fastq.gz
../data/reads/91.trimmed.R1.fastq.gz
../data/reads/91.trimmed.R2.fastq.gz
../data/reads/92.trimmed.R1.fastq.gz
../data/reads/92.trimmed.R2.fastq.gz
../data/reads/94.trimmed.R1.fastq.gz
../data/reads/94.trimmed.R2.fastq.gz
../data/reads/97.trimmed.R1.fastq.gz
../data/reads/97.trimmed.R2.fastq.gz
../data/reads/98.trimmed.R1.fastq.gz
../data/reads/98.trimmed.R2.fastq.gz
../data/reads/99.trimmed.R1.fastq.gz
../data/reads/99.trimmed.R2.fastq.gz
../data/reads/splice-test-files.txtNeed to find treatment file…
3 Genome
https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_031168955.1/
cd ../data
/home/shared/datasets download genome accession GCF_031168955.1 --include gff3,gtf,rna,cds,protein,genome,seq-reportcd ../data
unzip ncbi_dataset.zipls /home/shared/8TB_HDD_03/sr320/github/project-cod-temperature/data/ncbi_dataset/data/GCF_031168955.1cds_from_genomic.fna
GCF_031168955.1_ASM3116895v1_genomic.fna
genomic.gff
genomic.gtf
protein.faa
rna.fna
sequence_report.jsonl4 Hisat
/home/shared/hisat2-2.2.1/hisat2_extract_exons.py \
../data/ncbi_dataset/data/GCF_031168955.1/genomic.gtf \
> ../output/10-hisat-deseq2/m_exon.tab/home/shared/hisat2-2.2.1/hisat2_extract_splice_sites.py \
../data/ncbi_dataset/data/GCF_031168955.1/genomic.gtf \
> ../output/10-hisat-deseq2/m_spice_sites.tabecho "10-hisat-deseq2/GCF*" >> ../output/.gitignore/home/shared/hisat2-2.2.1/hisat2-build \
../data/ncbi_dataset/data/GCF_031168955.1/GCF_031168955.1_ASM3116895v1_genomic.fna \
../output/10-hisat-deseq2/GCF_031168955.1.index \
--exon ../output/10-hisat-deseq2/m_exon.tab \
--ss ../output/10-hisat-deseq2/m_spice_sites.tab \
-p 20 \
../data/ncbi_dataset/data/GCF_031168955.1/genomic.gtf \
2> ../output/10-hisat-deseq2/hisat2-build_stats.txtecho "10-hisat-deseq2/*sam" >> ../output/.gitignorefind ../data/reads/*.trimmed.R1.fastq.gz \
| xargs basename -s .trimmed.R1.fastq.gz | xargs -I{} \
/home/shared/hisat2-2.2.1/hisat2 \
-x ../output/10-hisat-deseq2/GCF_031168955.1.index \
--dta \
-p 20 \
-1 ../data/reads/{}.trimmed.R1.fastq.gz \
-2 ../data/reads/{}.trimmed.R2.fastq.gz \
-S ../output/10-hisat-deseq2/{}.sam \
2> ../output/10-hisat-deseq2/hisat.outecho "10-hisat-deseq2/*bam" >> ../output/.gitignore
echo "10-hisat-deseq2/*bam*" >> ../output/.gitignorefor samfile in ../output/10-hisat-deseq2/*.sam; do
bamfile="${samfile%.sam}.bam"
sorted_bamfile="${samfile%.sam}.sorted.bam"
/home/shared/samtools-1.12/samtools view -bS -@ 20 "$samfile" > "$bamfile"
/home/shared/samtools-1.12/samtools sort -@ 20 "$bamfile" -o "$sorted_bamfile"
/home/shared/samtools-1.12/samtools index -@ 20 "$sorted_bamfile"
donerm ../output/10-hisat-deseq2/*samls ../output/10-hisat-deseq2/*sorted.bam | wc -ltail ../output/10-hisat-deseq2/hisat.out ----
3794910 pairs aligned concordantly 0 times; of these:
146293 (3.85%) aligned discordantly 1 time
----
3648617 pairs aligned 0 times concordantly or discordantly; of these:
7297234 mates make up the pairs; of these:
5640288 (77.29%) aligned 0 times
1498025 (20.53%) aligned exactly 1 time
158921 (2.18%) aligned >1 times
82.81% overall alignment ratecat ../output/10-hisat-deseq2/hisat.out \
| grep "overall alignment rate"87.53% overall alignment rate
86.32% overall alignment rate
81.37% overall alignment rate
86.39% overall alignment rate
83.54% overall alignment rate
87.67% overall alignment rate
86.69% overall alignment rate
86.56% overall alignment rate
84.58% overall alignment rate
81.26% overall alignment rate
83.03% overall alignment rate
86.29% overall alignment rate
84.82% overall alignment rate
82.29% overall alignment rate
83.16% overall alignment rate
86.14% overall alignment rate
81.04% overall alignment rate
87.28% overall alignment rate
84.89% overall alignment rate
87.76% overall alignment rate
88.45% overall alignment rate
83.13% overall alignment rate
83.21% overall alignment rate
84.99% overall alignment rate
86.33% overall alignment rate
86.47% overall alignment rate
84.37% overall alignment rate
87.60% overall alignment rate
75.31% overall alignment rate
81.73% overall alignment rate
84.47% overall alignment rate
80.84% overall alignment rate
86.72% overall alignment rate
87.60% overall alignment rate
81.89% overall alignment rate
82.81% overall alignment rate5 Stringtie
echo "10-hisat-deseq2/*gtf" >> ../output/.gitignorefind ../output/10-hisat-deseq2/*sorted.bam \
| xargs basename -s .sorted.bam | xargs -I{} \
/home/shared/stringtie-2.2.1.Linux_x86_64/stringtie \
-p 36 \
-eB \
-G ../data/ncbi_dataset/data/GCF_031168955.1/genomic.gff \
-o ../output/10-hisat-deseq2/{}.gtf \
../output/10-hisat-deseq2/{}.sorted.bam6 Count Matrix
list format
RNA-ACR-140 ../output/15-Apul-hisat/RNA-ACR-140.gtf
RNA-ACR-145 ../output/15-Apul-hisat/RNA-ACR-145.gtf
RNA-ACR-173 ../output/15-Apul-hisat/RNA-ACR-173.gtf
RNA-ACR-178 ../output/15-Apul-hisat/RNA-ACR-178.gtfls ../output/10-hisat-deseq2/*gtf../output/10-hisat-deseq2/100.gtf
../output/10-hisat-deseq2/107.gtf
../output/10-hisat-deseq2/108.gtf
../output/10-hisat-deseq2/109.gtf
../output/10-hisat-deseq2/10.gtf
../output/10-hisat-deseq2/110.gtf
../output/10-hisat-deseq2/116.gtf
../output/10-hisat-deseq2/11.gtf
../output/10-hisat-deseq2/12.gtf
../output/10-hisat-deseq2/13.gtf
../output/10-hisat-deseq2/18.gtf
../output/10-hisat-deseq2/19.gtf
../output/10-hisat-deseq2/1.gtf
../output/10-hisat-deseq2/20.gtf
../output/10-hisat-deseq2/21.gtf
../output/10-hisat-deseq2/28.gtf
../output/10-hisat-deseq2/29.gtf
../output/10-hisat-deseq2/2.gtf
../output/10-hisat-deseq2/30.gtf
../output/10-hisat-deseq2/31.gtf
../output/10-hisat-deseq2/36.gtf
../output/10-hisat-deseq2/3.gtf
../output/10-hisat-deseq2/4.gtf
../output/10-hisat-deseq2/5.gtf
../output/10-hisat-deseq2/78.gtf
../output/10-hisat-deseq2/79.gtf
../output/10-hisat-deseq2/80.gtf
../output/10-hisat-deseq2/83.gtf
../output/10-hisat-deseq2/88.gtf
../output/10-hisat-deseq2/90.gtf
../output/10-hisat-deseq2/91.gtf
../output/10-hisat-deseq2/92.gtf
../output/10-hisat-deseq2/94.gtf
../output/10-hisat-deseq2/97.gtf
../output/10-hisat-deseq2/98.gtf
../output/10-hisat-deseq2/99.gtfhead ../data/list01.txt1 ../output/10-hisat-deseq2/1.gtf
2 ../output/10-hisat-deseq2/2.gtf
3 ../output/10-hisat-deseq2/3.gtf
4 ../output/10-hisat-deseq2/4.gtf
5 ../output/10-hisat-deseq2/5.gtf
10 ../output/10-hisat-deseq2/10.gtf
11 ../output/10-hisat-deseq2/11.gtf
12 ../output/10-hisat-deseq2/12.gtf
13 ../output/10-hisat-deseq2/13.gtf
18 ../output/10-hisat-deseq2/18.gtfpython /home/shared/stringtie-2.2.1.Linux_x86_64/prepDE.py \
-i ../data/list01.txt \
-g ../output/10-hisat-deseq2/gene_count_matrix.csv \
-t ../output/10-hisat-deseq2/transcript_count_matrix.csvhead ../output/10-hisat-deseq2/*matrix.csv==> ../output/10-hisat-deseq2/gene_count_matrix.csv <==
gene_id,1,10,100,107,108,109,11,110,116,12,13,18,19,2,20,21,28,29,3,30,31,36,4,5,78,79,80,83,88,90,91,92,94,97,98,99
gene-LOC132462341|LOC132462341,360,464,391,346,691,408,436,509,366,373,432,385,330,288,307,346,293,347,452,452,984,230,469,400,472,311,368,312,551,631,605,366,339,517,577,235
gene-abce1|abce1,694,325,276,77,409,196,281,284,327,310,393,386,106,290,252,326,345,254,363,353,922,160,588,285,178,119,197,134,128,103,315,204,301,56,49,260
gene-si:dkey-6i22.5|si:dkey-6i22.5,0,10,13,11,49,42,14,36,0,14,9,0,0,14,3,21,26,9,39,20,10,0,0,10,48,41,60,39,49,54,20,27,7,22,63,12
gene-ube2v1|ube2v1,10,22,15,19,74,28,63,35,30,31,25,42,17,14,21,60,61,14,29,37,68,37,0,30,10,13,56,32,45,55,27,38,60,25,18,50
gene-cldn15la|cldn15la,0,9,0,0,18,0,0,0,0,27,0,0,0,0,0,0,0,0,4,35,0,0,0,4,0,0,0,0,8,32,0,0,14,0,0,0
gene-muc15|muc15,0,40,40,29,8,0,38,0,16,15,0,0,0,0,22,22,69,22,0,15,0,34,55,51,0,30,19,31,0,0,42,34,0,0,26,56
gene-pcloa|pcloa,0,3,0,0,41,7,4,0,3,0,0,0,27,0,0,0,0,4,0,10,0,23,0,0,0,0,4,4,0,0,4,0,0,22,101,0
gene-LOC132472829|LOC132472829,402,550,293,182,1082,213,234,149,444,360,531,242,269,194,359,383,395,392,408,489,310,352,168,403,462,287,310,113,457,321,396,234,455,279,229,208
gene-ifi35|ifi35,3696,2492,2615,2184,2702,2579,3631,3158,2121,2673,2893,2210,2324,2384,2561,2654,2887,2250,3143,2830,1297,1981,2971,2651,2669,2671,1790,2173,2603,3260,2186,2451,2464,2768,1935,1991
==> ../output/10-hisat-deseq2/transcript_count_matrix.csv <==
transcript_id,1,10,100,107,108,109,11,110,116,12,13,18,19,2,20,21,28,29,3,30,31,36,4,5,78,79,80,83,88,90,91,92,94,97,98,99
rna-XM_060037252.1,20,918,84,11,385,70,54,82,131,72,52,8,26,41,33,62,30,12,45,85,377,59,58,57,96,19,55,94,92,125,54,34,69,36,16,83
rna-XM_060070002.1,0,0,0,0,0,12,0,0,0,14,14,0,0,0,0,0,0,3,0,0,0,0,0,0,0,0,0,0,0,0,13,0,0,0,0,7
rna-XM_060056394.1,0,0,0,1,0,0,336,0,37,0,0,0,86,0,0,0,0,0,395,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,1
rna-XM_060072836.1,0,0,14,4,0,0,5,0,15,0,0,0,4,0,0,5,0,0,0,11,0,0,5,0,0,12,0,0,12,3,10,0,9,0,0,0
rna-XM_060062883.1,719,38,380,279,652,462,461,381,432,0,831,479,641,171,593,82,520,625,366,533,640,311,36,876,365,322,351,342,430,324,450,410,540,336,513,370
rna-XM_060057326.1,1072,1577,801,1056,1420,909,1523,1101,951,1459,2162,1170,1983,1317,1264,1398,2075,1055,1303,1820,1496,934,966,1509,1132,928,845,948,1930,2047,1039,781,979,2259,2747,740
rna-XM_060051796.1,0,1,1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,3,0,0,0,0,0,0,0,4,0,2,0,0,0,0,0,0,0,2
rna-XM_060076321.1,0,0,0,0,16,0,0,0,0,0,0,4,58,0,0,0,0,0,0,12,0,0,0,0,0,0,0,0,0,0,0,0,5,13,80,0
rna-XM_060064475.1,6,1771,834,10,33,0,1752,997,470,1572,0,0,16,713,0,0,0,5,0,679,292,656,0,1051,6,1326,0,0,0,0,592,448,0,0,0,57 DEseq2
need conditions.txt file in this format
SampleID Condition
RNA.ACR.140 control
RNA.ACR.145 control
RNA.ACR.173 treated
RNA.ACR.178 treatedhead ../data/conditions.txtSampleID Condition
X1 16
X2 16
X3 16
X4 16
X5 16
X10 16
X11 16
X12 16
X13 16library(DESeq2)# Load gene(/transcript) count matrix and labels
countData <- as.matrix(read.csv("../output/10-hisat-deseq2/gene_count_matrix.csv", row.names="gene_id"))
colData <- read.csv("../data/conditions.txt", sep="\t", row.names = 1)
# Note: The PHENO_DATA file contains information on each sample, e.g., sex or population.
# The exact way to import this depends on the format of the file.
# Check all sample IDs in colData are also in CountData and match their orders
all(rownames(colData) %in% colnames(countData)) # This should return TRUE[1] TRUEcountData <- countData[, rownames(colData)]
all(rownames(colData) == colnames(countData)) # This should also return TRUE[1] TRUE# Create a DESeqDataSet from count matrix and labels
dds <- DESeqDataSetFromMatrix(countData = countData,
colData = colData, design = ~ Condition)
# Run the default analysis for DESeq2 and generate results table
dds <- DESeq(dds)
deseq2.res <- results(dds)
# Sort by adjusted p-value and display
resOrdered <- deseq2.res[order(deseq2.res$padj), ]
vsd <- vst(dds, blind = FALSE)
plotPCA(vsd, intgroup = "Condition")
# Select top 50 differentially expressed genes
res <- results(dds)
res_ordered <- res[order(res$padj), ]
top_genes <- row.names(res_ordered)[1:50]
# Extract counts and normalize
counts <- counts(dds, normalized = TRUE)
counts_top <- counts[top_genes, ]
# Log-transform counts
log_counts_top <- log2(counts_top + 1)
# Generate heatmap
pheatmap(log_counts_top, scale = "row")
# Count number of hits with adjusted p-value less then 0.05
dim(res[!is.na(deseq2.res$padj) & deseq2.res$padj <= 0.05, ])[1] 5061 6tmp <- deseq2.res
# The main plot
plot(tmp$baseMean, tmp$log2FoldChange, pch=20, cex=0.45, ylim=c(-3, 3), log="x", col="darkgray",
main="DEG Dessication (pval <= 0.05)",
xlab="mean of normalized counts",
ylab="Log2 Fold Change")
# Getting the significant points and plotting them again so they're a different color
tmp.sig <- deseq2.res[!is.na(deseq2.res$padj) & deseq2.res$padj <= 0.05, ]
points(tmp.sig$baseMean, tmp.sig$log2FoldChange, pch=20, cex=0.45, col="red")
# 2 FC lines
abline(h=c(-1,1), col="blue")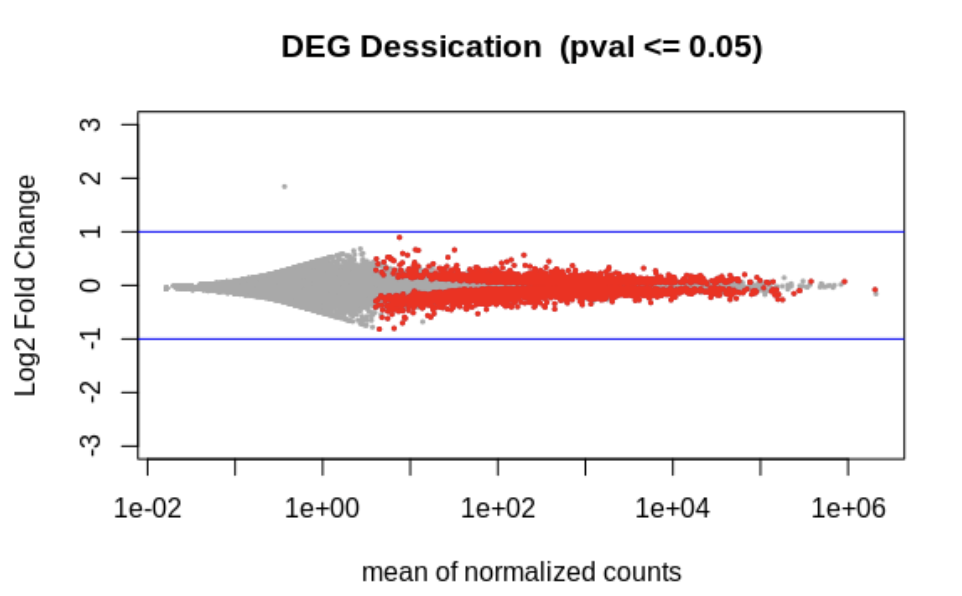
write.table(tmp.sig, "../output/10-hisat-deseq2/DEGlist.tab", sep = '\t', row.names = T)deglist <- read.csv("../output/10-hisat-deseq2/DEGlist.tab", sep = '\t', header = TRUE)
deglist$RowName <- rownames(deglist)
deglist2 <- deglist[, c("RowName", "pvalue")] # Optionally, reorder the columnshead(deglist) baseMean log2FoldChange lfcSE
gene-abce1|abce1 262.70027 0.10595278 0.02812204
gene-si:dkey-6i22.5|si:dkey-6i22.5 23.18182 -0.27199275 0.07237460
gene-LOC132463139|LOC132463139 264.89473 0.29450501 0.05965925
gene-prkaa1|prkaa1 3214.61286 -0.12678273 0.03404263
gene-snx27a|snx27a 1220.23705 -0.06307107 0.01399906
gene-LOC132467924|LOC132467924 197.52277 0.09450712 0.03203037
stat pvalue padj
gene-abce1|abce1 3.767607 1.648201e-04 1.695796e-03
gene-si:dkey-6i22.5|si:dkey-6i22.5 -3.758124 1.711919e-04 1.744561e-03
gene-LOC132463139|LOC132463139 4.936451 7.955683e-07 2.120046e-05
gene-prkaa1|prkaa1 -3.724235 1.959086e-04 1.933956e-03
gene-snx27a|snx27a -4.505379 6.625450e-06 1.259621e-04
gene-LOC132467924|LOC132467924 2.950547 3.172118e-03 1.713888e-02
RowName
gene-abce1|abce1 gene-abce1|abce1
gene-si:dkey-6i22.5|si:dkey-6i22.5 gene-si:dkey-6i22.5|si:dkey-6i22.5
gene-LOC132463139|LOC132463139 gene-LOC132463139|LOC132463139
gene-prkaa1|prkaa1 gene-prkaa1|prkaa1
gene-snx27a|snx27a gene-snx27a|snx27a
gene-LOC132467924|LOC132467924 gene-LOC132467924|LOC132467924