
head ../analyses/12-fix-gff/mod_augustus.gtfWhile there are still some questions related to overall alignment rates and sample specific alignment rates, it is possible that the mess of gtf might be overcome.
The problem seemed to have arisen based on the fact NCBI renamed chromosomes whereas annotations (sitting on Dryad) have different IDs. Using a table on NCBI and realizing from author the numbers in their IDs corresponded to chromosome number.
Making a new GTF
as there are quotes in original gtf I needed to add quote = ""
```{r}
gtfbttr <- read.table("../data/augustus.hints.gtf", sep = "\t", quote = "")
```added a new column with chromosome number
```{r}
gtfch <- gtfbttr %>%
mutate(Chromosome.name = str_extract(V1, "(?<=\\.)\\d+"), # Extract the number after the dot
Chromosome.name = str_remove_all(Chromosome.name, "^0+")) # Remove leading zeros
```now joining with NCBI table to get the GenBank.seq.accession
```{r}
gtfch %>%
left_join(ncbi, by = "Chromosome.name") %>%
select(GenBank.seq.accession, V2, V3, V4, V5, V6, V7, V8, V9) %>% filter(!is.na(GenBank.seq.accession)) %>%
write.table(file = "../analyses/12-fix-gff/mod_augustus.gtf", sep = "\t", quote = FALSE, row.names = FALSE, col.names = FALSE)
```this new gtf is in rpos and can be found here
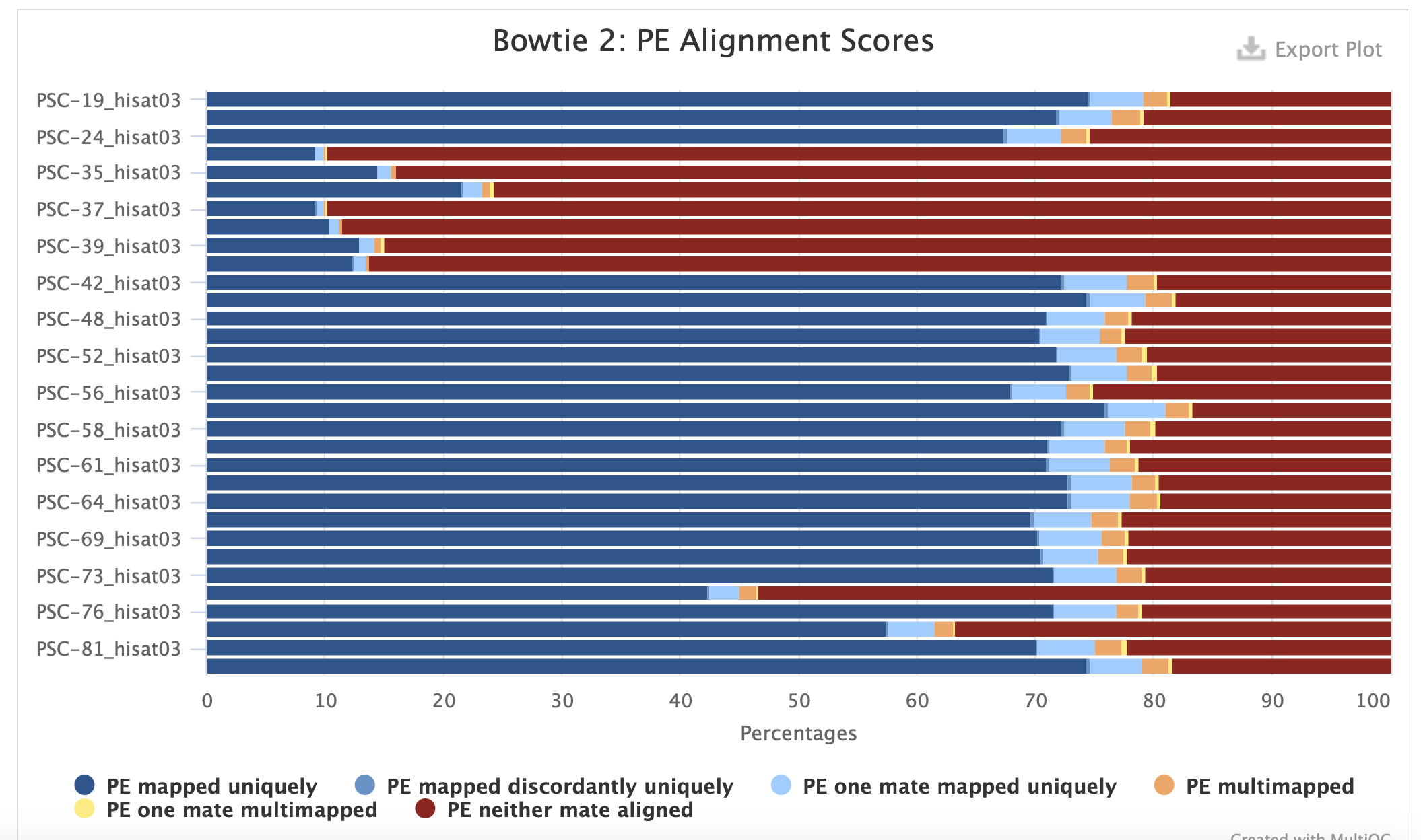
Reads
ls /home/shared/8TB_HDD_02/graceac9/data/pycno2021/*/home/shared/FastQC-0.12.1/fastqc \
/home/shared/8TB_HDD_02/graceac9/data/pycno2021/*fq.gz \
-t 36 \
-o ../analyses/13-hisat-deseq2/eval "$(/opt/anaconda/anaconda3/bin/conda shell.bash hook)"
conda activate
which multiqc
multiqc ../analyses/13-hisat-deseq2/ \
-o ../analyses/13-hisat-deseq2/Genome
https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_032158295.1/
cd ../data
/home/shared/datasets download genome accession GCA_032158295.1 --include gff3,rna,cds,protein,genome,seq-reportcd ../data
unzip ncbi_dataset.zipls ../data/ncbi_dataset/data/GCA_032158295.1Annotation files
head ../analyses/12-fix-gff/mod_augustus.gtf
head ../data/ncbi_dataset/data/GCA_032158295.1/GCA_032158295.1_ASM3215829v1_genomic.fnaHisat
/home/shared/hisat2-2.2.1/hisat2_extract_exons.py \
../analyses/12-fix-gff/mod_augustus.gtf \
> ../analyses/13-hisat-deseq2/m_exon.tab/home/shared/hisat2-2.2.1/hisat2_extract_splice_sites.py \
../analyses/12-fix-gff/mod_augustus.gtf \
> ../analyses/13-hisat-deseq2/m_spice_sites.tabecho "13-hisat-deseq2/GCF*" >> ../analyses/.gitignore
echo "13-hisat-deseq2/GCF**fastq" >> ../analyses/.gitignore/home/shared/hisat2-2.2.1/hisat2-build \
../data/ncbi_dataset/data/GCA_032158295.1/GCA_032158295.1_ASM3215829v1_genomic.fna \
../analyses/13-hisat-deseq2/GCA_032158295.index \
--exon ../analyses/13-hisat-deseq2/m_exon.tab \
--ss ../analyses/13-hisat-deseq2/m_spice_sites.tab \
-p 20 \
../analyses/12-fix-gff/mod_augustus.gtf \
2> ../analyses/13-hisat-deseq2/hisat2-build_stats.txtecho "13-hisat-deseq2/*sam" >> ../analyses/.gitignorefind /home/shared/8TB_HDD_02/graceac9/data/pycno2021/*_R1_001.fastq.gz.fastp-trim.20220810.fq.gz | xargs basename -s _R1_001.fastq.gz.fastp-trim.20220810.fq.gz | xargs -I{} echo {}keeping unmapped reads
find /home/shared/8TB_HDD_02/graceac9/data/pycno2021/*_R1_001.fastq.gz.fastp-trim.20220810.fq.gz \
| xargs -I{} basename -s _R1_001.fastq.gz.fastp-trim.20220810.fq.gz {} \
| xargs -I{} sh -c '/home/shared/hisat2-2.2.1/hisat2 \
-x ../analyses/13-hisat-deseq2/GCA_032158295.index \
--dta \
-p 32 \
-1 /home/shared/8TB_HDD_02/graceac9/data/pycno2021/{}_R1_001.fastq.gz.fastp-trim.20220810.fq.gz \
-2 /home/shared/8TB_HDD_02/graceac9/data/pycno2021/{}_R2_001.fastq.gz.fastp-trim.20220810.fq.gz \
-S ../analyses/13-hisat-deseq2/{}_03.sam \
--un-conc ../analyses/13-hisat-deseq2/{}_unmapped_reads.fastq \
> ../analyses/13-hisat-deseq2/{}_hisat03.stdout 2> ../analyses/13-hisat-deseq2/{}_hisat03.stderr'Explanation xargs -I{}: This option allows you to replace {} in the command with the output from the previous command (i.e., basename). It’s used twice: first, to strip the suffix from the filenames, and second, to construct and execute the hisat2 command.
sh -c: This is used to execute a complex command within xargs. It’s necessary because the output redirection (>, 2>) is shell functionality, and without sh -c, xargs wouldn’t handle it correctly.
echo "13-hisat-deseq2/*bam" >> ../analyses/.gitignore
echo "13-hisat-deseq2/*bam*" >> ../analyses/.gitignorefor samfile in ../analyses/13-hisat-deseq2/*.sam; do
bamfile="${samfile%.sam}.bam"
sorted_bamfile="${samfile%.sam}.sorted.bam"
/home/shared/samtools-1.12/samtools view -bS -@ 20 "$samfile" > "$bamfile"
/home/shared/samtools-1.12/samtools sort -@ 20 "$bamfile" -o "$sorted_bamfile"
/home/shared/samtools-1.12/samtools index -@ 20 "$sorted_bamfile"
done,
rm ../analyses/13-hisat-deseq2/*samls ../analyses/13-hisat-deseq2/*sorted.bam | wc -lStringtie
echo "13-hisat-deseq2/*gtf" >> ../analyses/.gitignore/home/shared/gffread-0.12.7.Linux_x86_64/gffread \
../analyses/12-fix-gff/mod_augustus.gtf \
-T \
-o ../analyses/13-hisat-deseq2/mod_augustus.gfffind ../analyses/13-hisat-deseq2/*sorted.bam \
| xargs basename -s .sorted.bam | xargs -I{} \
sh -c '/home/shared/stringtie-2.2.1.Linux_x86_64/stringtie \
-p 36 \
-eB \
-G ../analyses/13-hisat-deseq2/mod_augustus.gff \
-o ../analyses/13-hisat-deseq2/{}.gtf \
../analyses/13-hisat-deseq2/{}.sorted.bam'eval "$(/opt/anaconda/anaconda3/bin/conda shell.bash hook)"
conda activate
which multiqc
multiqc ../analyses/13-hisat-deseq2/ \
-o ../analyses/13-hisat-deseq2/