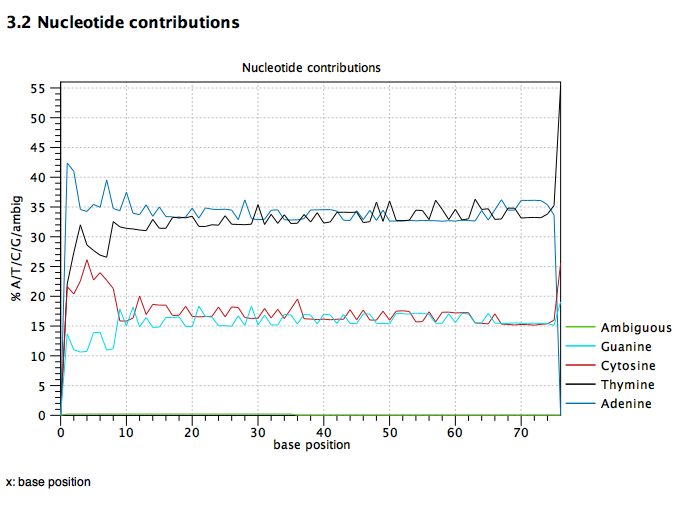
Clc And Virginica
It seems that mapping rates can very a lot. We have a new data set comparing WG-BS, RRBS, and MBDBS. This is valuable as it offers data to run the math on what is most optimal. This first step is mapping. Here I explore CLC results as we are working with Qiagen and this is the software the are using.
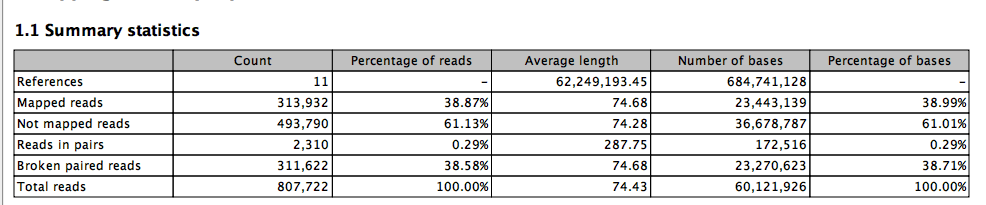
Lets start with default..
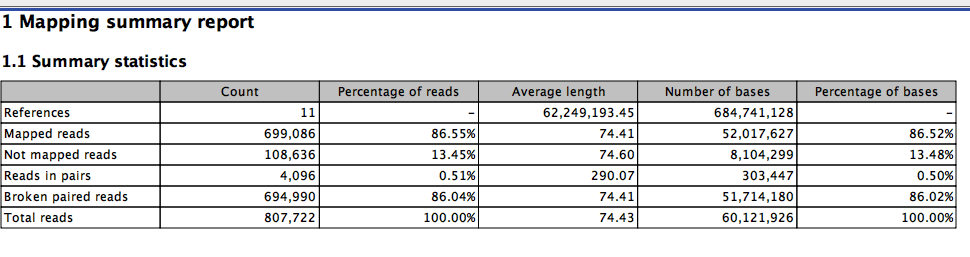
The full read
Map Bisulfite Reads to Reference Setting up the mapping of bisulfite sequencing reads to reference is no different from the usual read mapping, see for explanation of inputs, parameters and outputs. Some options, like ‘Global alignment’, that are not generally advisable to change for bisulfite mapping are not available, or are preset. In particular: only a track version of a reference sequence is accepted as input for mapping; only a track version of mappings is produced in output. The bisulfite mappings in CLC Workbenches have a special ‘invisible’ property set for them, to inform the downstream tool Call Methylation levels (See section 1.3) about correct type of input. An optional mapping report has the same sections as the regular one. Please note that, because two versions of the reference sequence (C->T, and G->A - converted) have to be indexed and used simultaneously for each read, the RAM requirements for the bisulfite mapper are double of the regular one, for the same-size reference sequence.

Figure 1.4: A typical directional shotgun BS-seq mapping, together with the base-level methylation calling feature track on top. Fig. 1.4 illustrates the view of a typical directional shotgun BS-seq mapping. As with any read mapping view, the colour of the reads follows the usual CLC convention, that is green/red for forward/reverse reads, and dark/pale blue for paired reads. Independent of this orientation property, each read, or read pair has an ‘invisible’ property indicating if it came from the original top (OT), or original bottom (OB) strand. However, if the BS-sequencing protocol is truly 100%-directional, the orientation in the mapping, and the OT/OB origin of reads/read pairs will be concordant. In this figure, blocks of reads from the original top strand are marked with squiggly brackets on the left, the rest are from the original bottom strand. In a mapping, they can be distinguished by a pattern of highlighted mismatches to reference. OT reads will have mismatches predominantly on ‘T’, due to C->T conversion; OB reads will have a pattern of mismatches on ‘A’ symbols, corresponding to G->A conversion, as seen on a reverse-complementary strand. When methyl-C occurs in a sample, there will be a match in the reads, instead of an expected mismatch. In this figure, there were two positions where such events occurred, both on the original bottom strand, and both supported by a single read only. ‘G’ symbols in those reads are shown in red boxes. The reverse direction of an arrowhead on a base-level methylation track also reflects the OB-position of a methylation event. Note that in this example it appears that in the second position there may also be a G/T heterozygous SNP (C/A in the OB strand). While such occurrences may lead to underestimation of true methylation levels of cytosine alleles in heterozygous SNPs, our current tool does not attempt to compensate for those eventualities. To understand and interpret BS-sequencing and mapping better, it may be helpful to examine the position (marked with a red asterisk) in between the two detected methylation events. There appears to be an additional A/C heterozygous SNP with C’s in reads from OT strand fully converted to T’s, i.e. showing no evidence of methylation at that heterozygous position.
Now with a tweak..
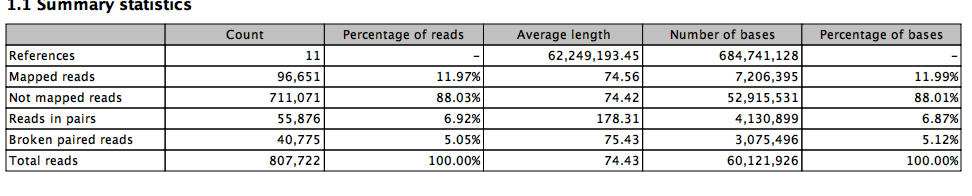
Big Difference.
third time
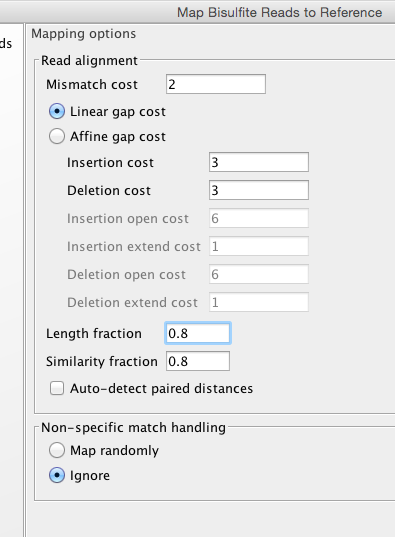
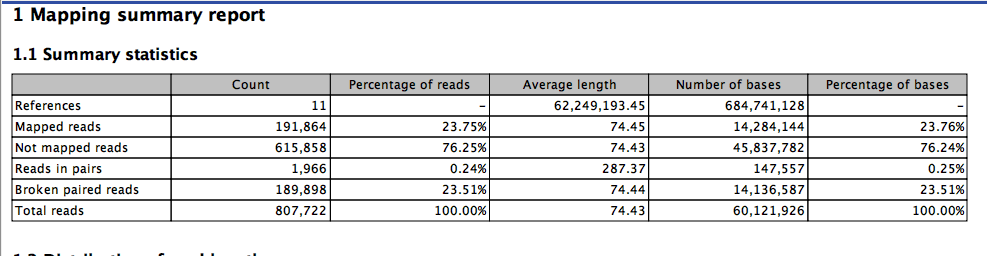
and one more.
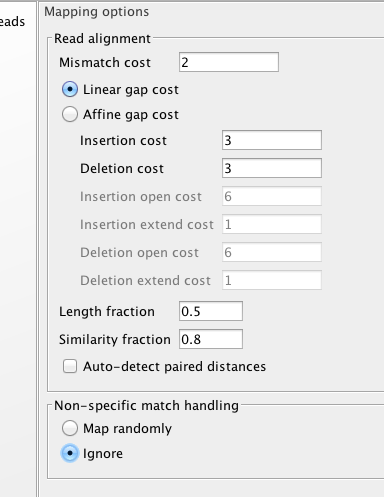
Next up maybe trim 5’ and see what happens