
Get in Control - Cv multiomics
In an effort to couple DNA methylation data to complementary RNA-seq data we are looking at what the DNA methylation landscape, DML look like. Oysters were exposed to ocean acidification. Males and females were included.
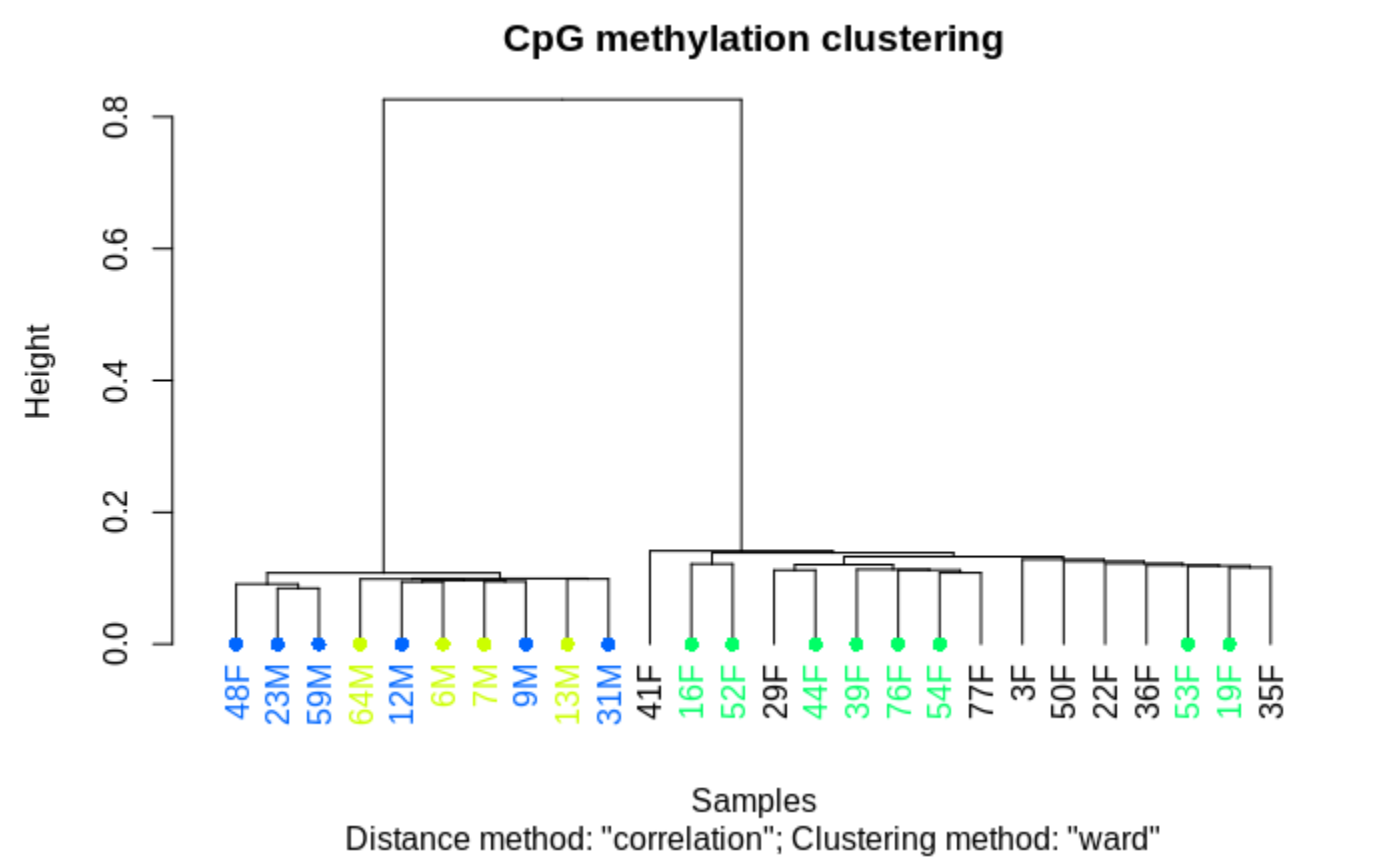
TLDR- Sex related methylation differences are FAR greater than any OA associated methylation differences.
Sample information
| Sample.ID | OldSample.ID | Treatment | Sex | TreatmentN | Parent.ID |
|---|---|---|---|---|---|
| 12M | S12M | Exposed | M | 3 | EM05 |
| 13M | S13M | Control | M | 1 | CM04 |
| 16F | S16F | Control | F | 2 | CF05 |
| 19F | S19F | Control | F | 2 | CF08 |
| 22F | S22F | Exposed | F | 4 | EF02 |
| 23M | S23M | Exposed | M | 3 | EM04 |
| 29F | S29F | Exposed | F | 4 | EF07 |
| 31M | S31M | Exposed | M | 3 | EM06 |
| 35F | S35F | Exposed | F | 4 | EF08 |
| 36F | S36F | Exposed | F | 4 | EF05 |
| 39F | S39F | Control | F | 2 | CF06 |
| 3F | S3F | Exposed | F | 4 | EF06 |
| 41F | S41F | Exposed | F | 4 | EF03 |
| 44F | S44F | Control | F | 2 | CF03 |
| 48M | S48M | Exposed | M | 3 | EM03 |
| 50F | S50F | Exposed | F | 4 | EF01 |
| 52F | S52F | Control | F | 2 | CF07 |
| 53F | S53F | Control | F | 2 | CF02 |
| 54F | S54F | Control | F | 2 | CF01 |
| 59M | S59M | Exposed | M | 3 | EM01 |
| 64M | S64M | Control | M | 1 | CM05 |
| 6M | S6M | Control | M | 1 | CM02 |
| 76F | S76F | Control | F | 2 | CF04 |
| 77F | S77F | Exposed | F | 4 | EF04 |
| 7M | S7M | Control | M | 1 | CM01 |
| 9M | S9M | Exposed | M | 3 | EM02 |
Looking just at controls
file.list_c=list('../bg_data/13M_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/16F_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/19F_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/39F_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/44F_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/52F_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/53F_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/54F_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/64M_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/6M_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/76F_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/7M_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam'
)
myobj_c = processBismarkAln(location = file.list_c,
sample.id = list("13M","16F","19F","39F","44F","52F","53F","54F", "64M", "6M", "76F", "7M"),
assembly = "cv",
read.context="CpG",
mincov=2,
treatment = c(0,1,1,1,1,1,1,1,0,0,1,0))
filtered.myobj=filterByCoverage(myobj_c,lo.count=10,lo.perc=NULL,
hi.count=NULL,hi.perc=98)
meth_filter=unite(filtered.myobj, min.per.group=NULL, destrand=TRUE)
clusterSamples(meth_filter, dist="correlation", method="ward", plot=TRUE)
PCASamples(meth_filter)
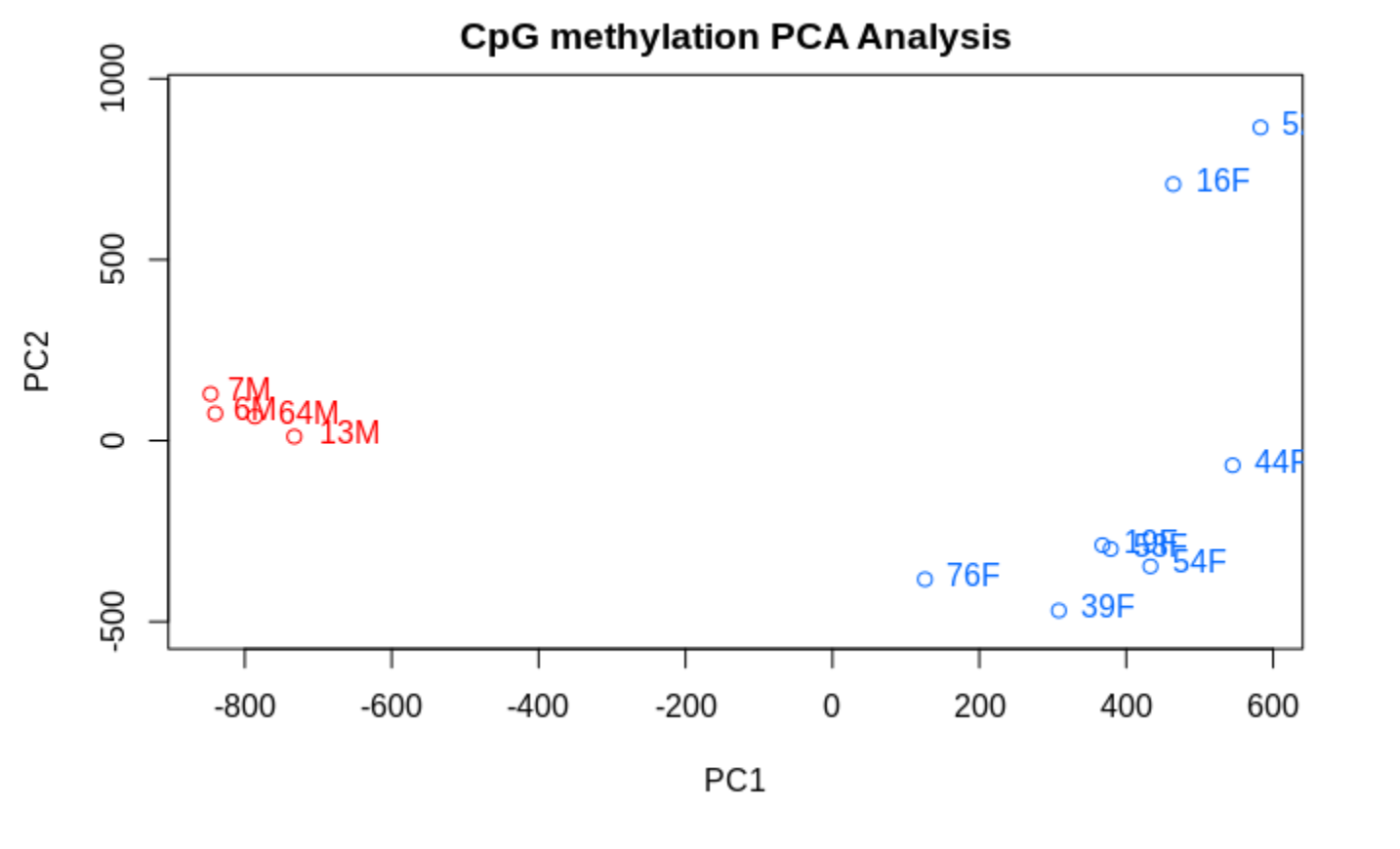
and when one includes
all samples
note different file list
file.list_all=list('../bg_data/12M_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/13M_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/16F_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/19F_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/22F_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/23M_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/29F_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/31M_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/35F_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/36F_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/39F_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/3F_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/41F_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/44F_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/48M_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/50F_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/52F_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/53F_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/54F_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/59M_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/64M_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/6M_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/76F_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/77F_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/7M_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam',
'../bg_data/9M_R1_val_1_bismark_bt2_pe.deduplicated.sorted.bam'
)
and align
myobj_all = processBismarkAln(location = file.list_all,
sample.id = list("12M","13M","16F","19F","22F","23M","29F","31M", "35F","36F","39F","3F","41F","44F","48M","50F","52F","53F","54F","59M","64M","6M","76F", "77F","7M","9M"),
assembly = "cv",
read.context="CpG",
mincov=2,
treatment = c(3,1,2,2,4,3,4,3,4,4,2,4,4,2,3,4,2,2,2,3,1,1,2,4,1,3))
Written on November 3, 2021