
Getting back to Oly
Visiting the long ago Oly WBGS data. Will start with see if can simply reproduce.
First will need to get trimmed reads onto the Mox.
reads present and launched
#!/bin/bash
## Job Name
#SBATCH --job-name=olywgbs
## Allocation Definition
#SBATCH --account=coenv
#SBATCH --partition=coenv
## Nodes
#SBATCH --nodes=1
## Walltime (days-hours:minutes:seconds format)
#SBATCH --time=5-00:00:00
## Memory per node
#SBATCH --mem=100G
#SBATCH --mail-type=ALL
#SBATCH --mail-user=sr320@uw.edu
## Specify the working directory for this job
#SBATCH --chdir=/gscratch/scrubbed/sr320/060321-oly
# Directories and programs
bismark_dir="/gscratch/srlab/programs/Bismark-0.22.3"
bowtie2_dir="/gscratch/srlab/programs/bowtie2-2.3.4.1-linux-x86_64/"
samtools="/gscratch/srlab/programs/samtools-1.9/samtools"
reads_dir="/gscratch/srlab/sr320/data/olurida-bs/"
genome_folder="/gscratch/srlab/sr320/data/olurida-bs/genome/"
source /gscratch/srlab/programs/scripts/paths.sh
${bismark_dir}/bismark_genome_preparation \
--verbose \
--parallel 28 \
--path_to_aligner ${bowtie2_dir} \
${genome_folder}
#1_ATCACG_L001_R1_001_trimmed.fq.gz
find ${reads_dir}*_R1_001_trimmed.fq.gz \
| xargs basename -s _R1_001_trimmed.fq.gz | xargs -I{} ${bismark_dir}/bismark \
--path_to_bowtie ${bowtie2_dir} \
-genome ${genome_folder} \
-p 4 \
-score_min L,0,-0.6 \
--non_directional \
${reads_dir}{}_R1_001_trimmed.fq.gz
find *.bam | \
xargs basename -s .bam | \
xargs -I{} ${bismark_dir}/deduplicate_bismark \
--bam \
--single \
{}.bam
${bismark_dir}/bismark_methylation_extractor \
--bedGraph --counts --scaffolds \
--multicore 28 \
--buffer_size 75% \
*deduplicated.bam
# Bismark processing report
${bismark_dir}/bismark2report
#Bismark summary report
${bismark_dir}/bismark2summary
#run multiqc
/gscratch/srlab/programs/anaconda3/bin/multiqc .
# Sort files for methylkit and IGV
find *deduplicated.bam | \
xargs basename -s .bam | \
xargs -I{} ${samtools} \
sort --threads 28 {}.bam \
-o {}.sorted.bam
# Index sorted files for IGV
# The "-@ 16" below specifies number of CPU threads to use.
find *.sorted.bam | \
xargs basename -s .sorted.bam | \
xargs -I{} ${samtools} \
index -@ 28 {}.sorted.bam
find *deduplicated.bismark.cov.gz \
| xargs basename -s _bismark_bt2_pe.deduplicated.bismark.cov.gz \
| xargs -I{} ${bismark_dir}/coverage2cytosine \
--genome_folder ${genome_folder} \
-o {} \
--merge_CpG \
--zero_based \
{}_bismark_bt2_pe.deduplicated.bismark.cov.gz
#creating bedgraphs post merge
for f in *merged_CpG_evidence.cov
do
STEM=$(basename "${f}" .CpG_report.merged_CpG_evidence.cov)
cat "${f}" | awk -F $'\t' 'BEGIN {OFS = FS} {if ($5+$6 >= 10) {print $1, $2, $3, $4}}' \
> "${STEM}"_10x.bedgraph
done
for f in *merged_CpG_evidence.cov
do
STEM=$(basename "${f}" .CpG_report.merged_CpG_evidence.cov)
cat "${f}" | awk -F $'\t' 'BEGIN {OFS = FS} {if ($5+$6 >= 5) {print $1, $2, $3, $4}}' \
> "${STEM}"_5x.bedgraph
done
#creating tab files with raw count for glms
for f in *merged_CpG_evidence.cov
do
STEM=$(basename "${f}" .CpG_report.merged_CpG_evidence.cov)
cat "${f}" | awk -F $'\t' 'BEGIN {OFS = FS} {if ($5+$6 >= 10) {print $1, $2, $3, $4, $5, $6}}' \
> "${STEM}"_10x.tab
done
for f in *merged_CpG_evidence.cov
do
STEM=$(basename "${f}" .CpG_report.merged_CpG_evidence.cov)
cat "${f}" | awk -F $'\t' 'BEGIN {OFS = FS} {if ($5+$6 >= 5) {print $1, $2, $3, $4, $5, $6}}' \
> "${STEM}"_5x.tab
done
The run is done..
Seem to have some issues
CpG_OT_8_ACTTGA_L001_R1_001_trimmed_bismark_bt2.deduplicated.txt
job.sh
*merged_CpG_evidence.cov_10x.bedgraph
*merged_CpG_evidence.cov_10x.tab
*merged_CpG_evidence.cov_5x.bedgraph
*merged_CpG_evidence.cov_5x.tab
multiqc_data
multiqc_report.html
slurm-1958552.out
Problem! is pe
find *deduplicated.bismark.cov.gz \
| xargs basename -s _bismark_bt2_pe.deduplicated.bismark.cov.gz \
| xargs -I{} ${bismark_dir}/coverage2cytosine \
--genome_folder ${genome_folder} \
-o {} \
--merge_CpG \
--zero_based \
{}_bismark_bt2_pe.deduplicated.bismark.cov.gz
Thus will run script that is just …
# Directories and programs
bismark_dir="/gscratch/srlab/programs/Bismark-0.22.3"
bowtie2_dir="/gscratch/srlab/programs/bowtie2-2.3.4.1-linux-x86_64/"
samtools="/gscratch/srlab/programs/samtools-1.9/samtools"
reads_dir="/gscratch/srlab/sr320/data/olurida-bs/"
genome_folder="/gscratch/srlab/sr320/data/olurida-bs/genome/"
source /gscratch/srlab/programs/scripts/paths.sh
find *deduplicated.bismark.cov.gz \
| xargs basename -s _L001_R1_001_trimmed_bismark_bt2.deduplicated.bismark.cov.gz \
| xargs -I{} ${bismark_dir}/coverage2cytosine \
--genome_folder ${genome_folder} \
-o {} \
--merge_CpG \
--zero_based \
{}_L001_R1_001_trimmed_bismark_bt2.deduplicated.bismark.cov.gz
#creating bedgraphs post merge
for f in *merged_CpG_evidence.cov
do
STEM=$(basename "${f}" .CpG_report.merged_CpG_evidence.cov)
cat "${f}" | awk -F $'\t' 'BEGIN {OFS = FS} {if ($5+$6 >= 10) {print $1, $2, $3, $4}}' \
> "${STEM}"_10x.bedgraph
done
for f in *merged_CpG_evidence.cov
do
STEM=$(basename "${f}" .CpG_report.merged_CpG_evidence.cov)
cat "${f}" | awk -F $'\t' 'BEGIN {OFS = FS} {if ($5+$6 >= 5) {print $1, $2, $3, $4}}' \
> "${STEM}"_5x.bedgraph
done
#creating tab files with raw count for glms
for f in *merged_CpG_evidence.cov
do
STEM=$(basename "${f}" .CpG_report.merged_CpG_evidence.cov)
cat "${f}" | awk -F $'\t' 'BEGIN {OFS = FS} {if ($5+$6 >= 10) {print $1, $2, $3, $4, $5, $6}}' \
> "${STEM}"_10x.tab
done
for f in *merged_CpG_evidence.cov
do
STEM=$(basename "${f}" .CpG_report.merged_CpG_evidence.cov)
cat "${f}" | awk -F $'\t' 'BEGIN {OFS = FS} {if ($5+$6 >= 5) {print $1, $2, $3, $4, $5, $6}}' \
> "${STEM}"_5x.tab
done
Now I think that fixed it.. will move off to gannet with a
sr320@Gannet:~$ cd /volume2/web/seashell/
sr320@Gannet:/volume2/web/seashell$ cd bu-mox/
sr320@Gannet:/volume2/web/seashell/bu-mox$ rsync -avz --exclude '*_to_*' --exclude 'CHG_*.txt' --exclude 'CHH_*.txt' --exclude 'CpG_*txt' --progress sr320@mox.hyak.uw.edu:/gscratch/scrubbed/sr320/ scrubbed/
Password:
What does it look like?
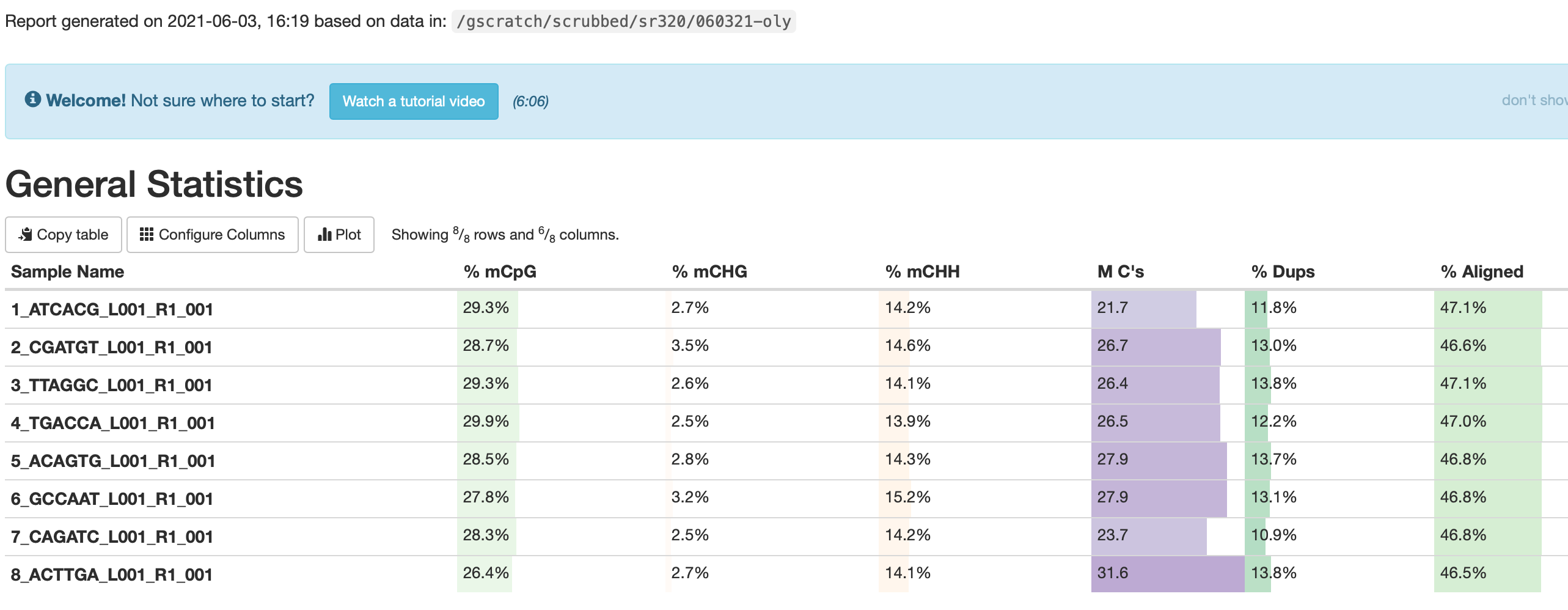
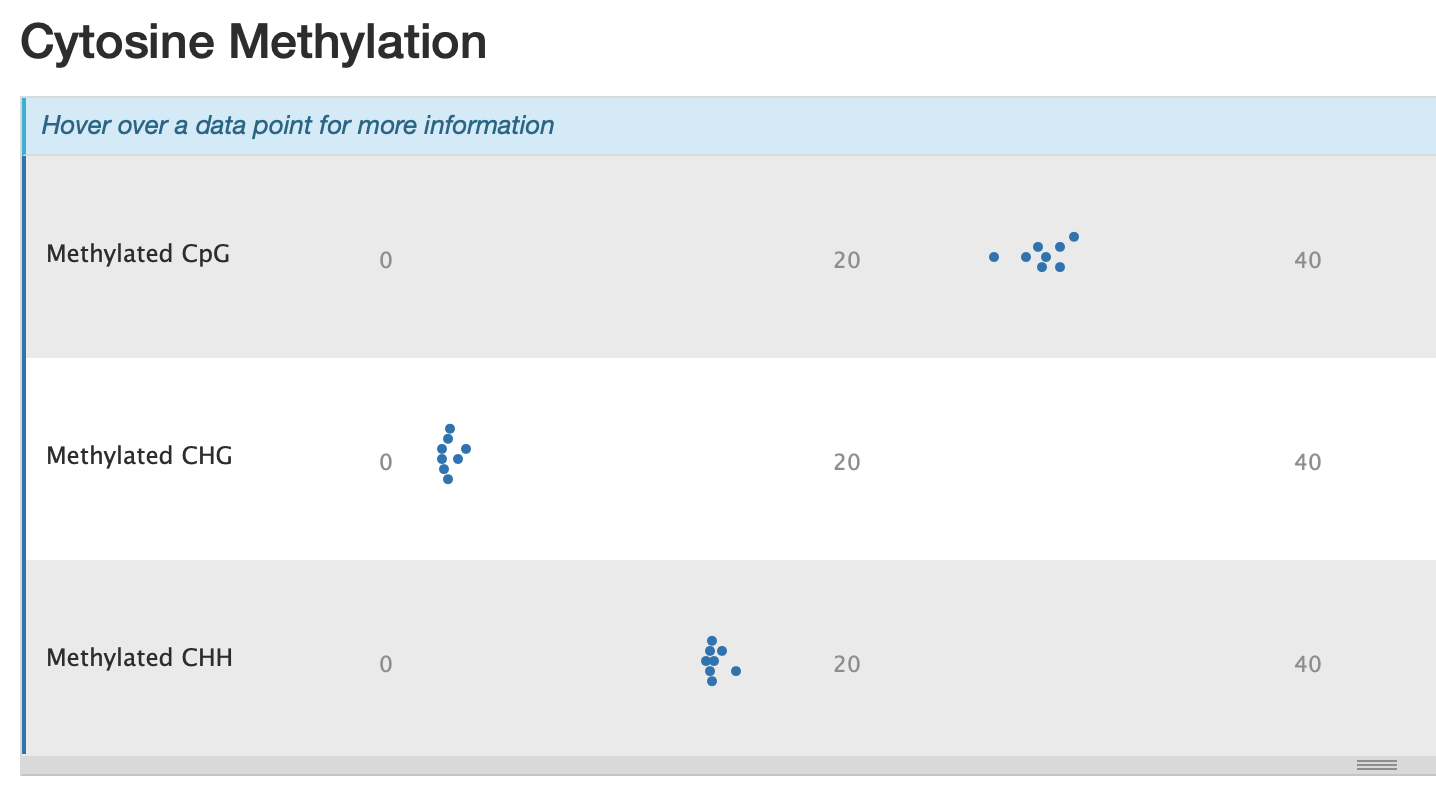
Oddly we have 30% CpG methylation and 14% CHH methylation..
Lets run a concatentated version..
script - https://d.pr/n/M6kCcF
All the output
-
individual - https://gannet.fish.washington.edu/seashell/bu-mox/scrubbed/060321-oly/
-
concat - https://gannet.fish.washington.edu/seashell/bu-mox/scrubbed/061021-oly/
lets go deeper on Concat..
https://d.pr/n/LIiTyU
in short
find ${reads_dir}combi* \
| xargs basename -s .fq.gz | xargs -I{} ${bismark_dir}/bismark \
--path_to_bowtie ${bowtie2_dir} \
-genome ${genome_folder} \
-p 4 \
-score_min L,0,-0.6 \
${reads_dir}{}.fq.gz
find *.bam | \
xargs basename -s .bam | \
xargs -I{} ${bismark_dir}/deduplicate_bismark \
--bam \
--single \
{}.bam
${bismark_dir}/bismark_methylation_extractor \
--single-end \
--comprehensive \
--merge_non_CpG \
--bedGraph \
--counts \
--scaffolds \
--multicore 28 \
--buffer_size 75% \
--report \
*deduplicated.bam
interesting we lost the non-cpg methylation

Now will go back and rerun individuals
script - https://d.pr/n/MXEg8s
Written on June 3, 2021