
Remember the five basic rules of database structure
- Order doesn’t matter
- No duplicate rows
- Every cell contains one value
- One column per type of information
- No redundant information
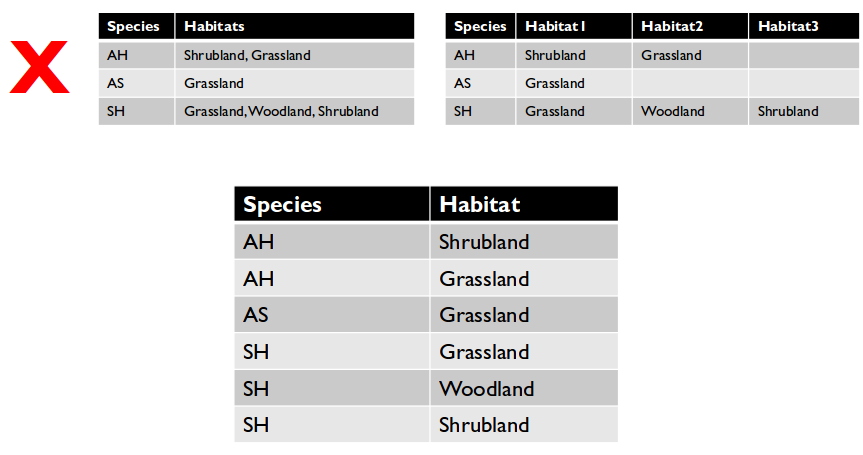
Restructure tables with messy data
- Cells with multiple values break rule #3.
- Redundant column information or cross-tabulated data breaks rule #4.
tidyr
library(tidyr)
- Package to deal with messy data
- Part of tidyverse
-
Gets data in format that works with ggplot2, dplyr, etc.
- Here is another messy dataset
- In wide format
genes_wide = data.frame(
name = c("A", "B", "C"),
a = c("16-Y", "25-N", "13-Y"),
t = c("1-N", "12-Y", "31-Y")
)
> genes_wide
name a t
1 A 16-Y 1-N
2 B 25-N 12-Y
3 C 13-Y 31-Y
- What do the values in the table represent?
A,B, andCare names of gene names16-Y,1-N, etc. represent:- Counts of bases
aandt - If a particular sequence involving that base is present in the gene,
YorN
- Counts of bases
Ask students,
- “What makes
genes_widemessy?”- “What are the variables in
genes_wide?”
- Tidy variables in
genes_widenameA,B, andC
baseaandt
base_counts- count
sequenceYorN
tidyr helps restructure messy data
gather()- Removes redundant columns
- Arguments:
- Piped
data.frame - Column name for grouping of old column headers
- Column name for grouping of old column values
- Column range for old columns with values
- Piped
- Gets data in long format
genes_long = genes_wide %>%
gather(base, base_counts, a:t)
> genes_long
name base base_counts
1 A a 16-Y
2 B a 25-N
3 C a 13-Y
4 A t 1-N
5 B t 12-Y
6 C t 31-Y
separate()- Separates multiple values in single column
- Arguments:
- Piped
data.frame - Column name
- New column names
- Separator value or character
- Piped
genes = genes_long %>%
separate(base_counts, c("counts", "sequence"), sep = "-")
> genes
name base counts sequence
1 A a 16 Y
2 B a 25 N
3 C a 13 Y
4 A t 1 N
5 B t 12 Y
6 C t 31 Y
- Use
spread()to turn data from long to wide format