Taking a set of unknown sequence files and annotating them
Screen Recording
For the first task you will take an unknown multi-fasta file and annotate it using blast. You are welcome to do this in terminal, Rstudio, or jupyter. My recommendation, and how I will demonstrate is using Rmarkdown. Once you have have your project structured, we will download software, databases, a fasta file and run the code.
Assignment
Annotate a provided fasta file with GO slim terms. Your code should be fully reproducible.
In your code directory create a file.
01-blast.Rmd
Tip
Rmarkdown is a good option as you can use markdown, add pictures and more!
cd /Applications/bioinfo/curl-O https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/ncbi-blast-2.13.0+-x64-macosx.tar.gztar-xf ncbi-blast-2.13.0+-x64-macosx.tar.gz
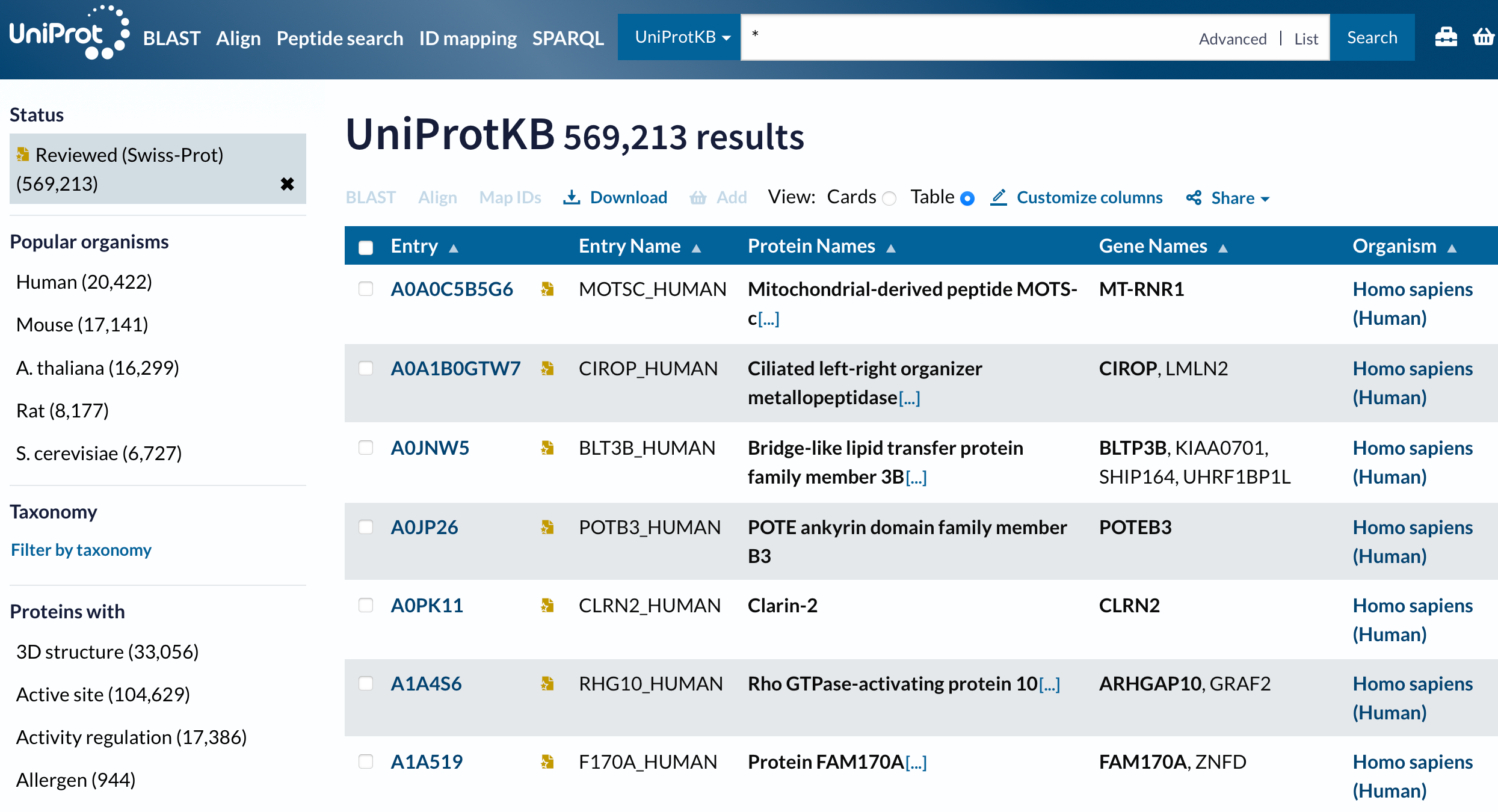
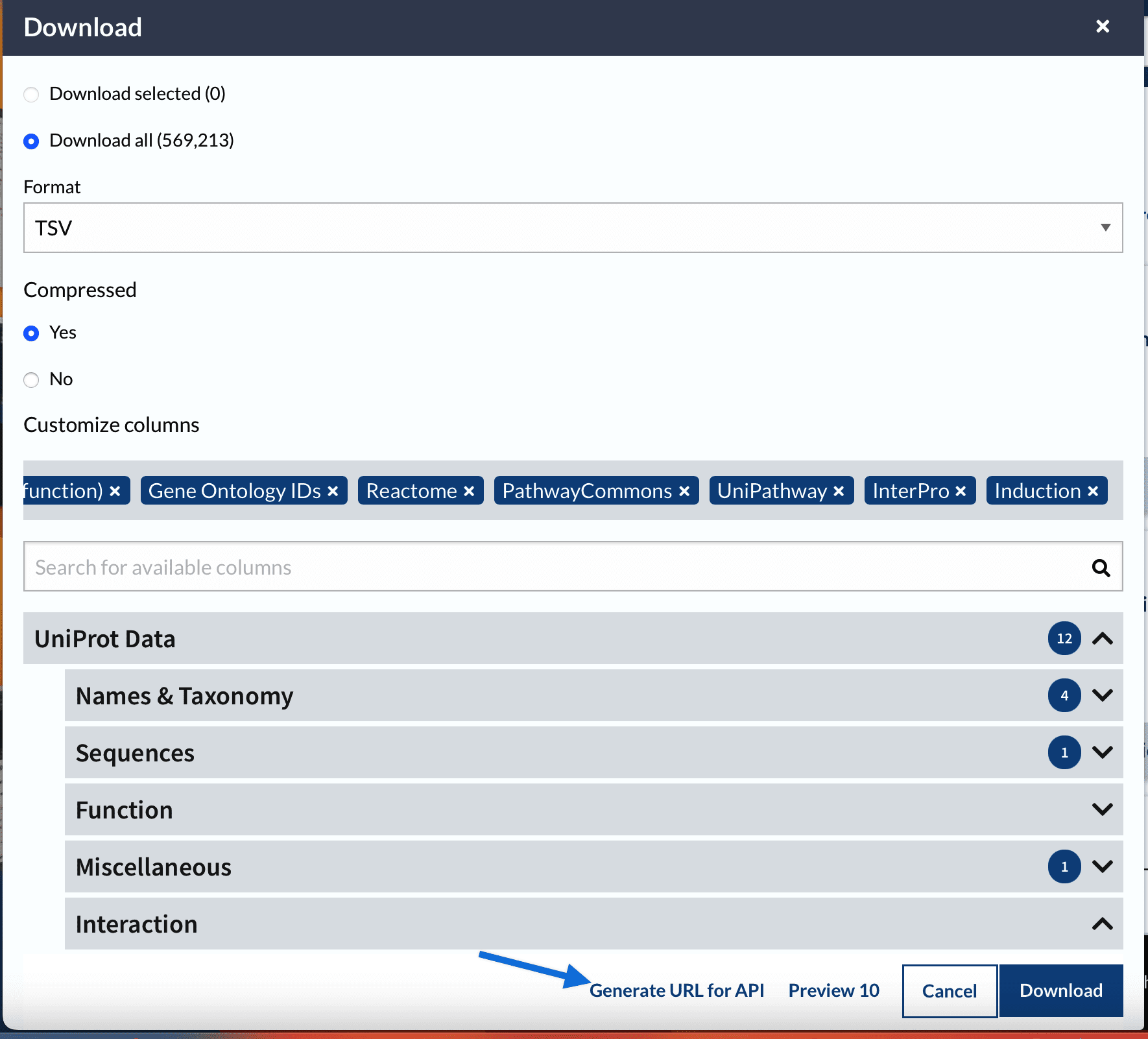
At this point we have a blast output table and annotation table both with a Uniprot accession number. Thus we can join the two tables and be able to get more functional information about thet genes.
---title: "NCBI Blast"subtitle: "Taking a set of unknown sequence files and annotating them"format: html: code-fold: false code-tools: true code-copy: true highlight-style: github code-overflow: wrap---## Screen Recording[{fig-align="left"}](https://washington.zoom.us/rec/share/r2_bDwXOIWDMG7_1WDr5PErr2RrwAnV6U-WG3giCxGRT-kL6fq7574awH38F4nw.2ayImTAVVNW1Er7G?startTime=1743696993000)For the first task you will take an unknown multi-fasta file and annotate it using blast. You are welcome to do this in terminal, Rstudio, or jupyter. My recommendation, and how I will demonstrate is using Rmarkdown. Once you have have your project structured, we will download software, databases, a fasta file and run the code.::: callout-important## AssignmentAnnotate a provided fasta file with GO slim terms. Your code should be fully reproducible.:::In your code directory create a file.`01-blast.Rmd`::: callout-tipRmarkdown is a good option as you can use markdown, add pictures and more!:::# Downloading softwareNCBI Blast Software is at<https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/>::: callout-tipIt is best to decide on central location on computer where software will be downloaded.:::https://github.com/RobertsLab/code/blob/master/09-blast.ipynb``` {.bash .code-overflow-wrap}cd /Applications/bioinfo/curl -O https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/ncbi-blast-2.13.0+-x64-macosx.tar.gztar -xf ncbi-blast-2.13.0+-x64-macosx.tar.gz`````` bashls /Applications/bioinfo/``````{bash}/Applications/bioinfo/ncbi-blast-2.13.0+/bin/blastx -h```# Make Blast Databasesee <https://www.uniprot.org/downloads>https://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/complete/uniprot_sprot.fasta.gz``` bashcd ../datacurl-O https://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/complete/uniprot_sprot.fasta.gzmv uniprot_sprot.fasta.gz uniprot_sprot_r2023_01.fasta.gzgunzip-k uniprot_sprot_r2023_01.fasta.gzls ../data`````` bash/Applications/bioinfo/ncbi-blast-2.13.0+/bin/makeblastdb\-in ../data/uniprot_sprot_r2023_01.fasta \-dbtype prot \-out ../blastdb/uniprot_sprot_r2023_01```# Get the query sequence``` bashcurl https://eagle.fish.washington.edu/cnidarian/Ab_4denovo_CLC6_a.fa \-k \> ../data/Ab_4denovo_CLC6_a.fa``````{bash}head ../data/Ab_4denovo_CLC6_a.faecho"How many sequences are there?"grep-c">" ../data/Ab_4denovo_CLC6_a.fa```# Run Blast``` bash/Applications/bioinfo/ncbi-blast-2.13.0+/bin/blastx\-query ../data/Ab_4denovo_CLC6_a.fa \-db ../blastdb/uniprot_sprot_r2023_01 \-out ../output/Ab_4-uniprot_blastx.tab \-evalue 1E-20 \-num_threads 20 \-max_target_seqs 1 \-outfmt 6``````{bash}head-2 ../output/Ab_4-uniprot_blastx.tabwc-l ../output/Ab_4-uniprot_blastx.tab```Need to convert `sp|Q08013|SSRG_RAT` to get accession number out.# Getting more information```{r schemat, echo = FALSE, out.width = "70%", fig.align = "center"}knitr::include_graphics("../img/uniprot.png")```https://www.uniprot.org/uniprotkb```{r api, echo = FALSE, out.width = "70%", fig.align = "center"}knitr::include_graphics("../img/uniprot-api.png")``````` ```{bash}curl -O -H "Accept: text/plain; format=tsv" "https://rest.uniprot.org/uniprotkb/stream?compressed=true&fields=accession%2Creviewed%2Cid%2Cprotein_name%2Cgene_names%2Corganism_name%2Clength%2Cgo_f%2Cgo%2Cgo_p%2Cgo_c%2Cgo_id%2Ccc_interaction%2Cec%2Cxref_reactome%2Cxref_unipathway%2Cxref_interpro&format=tsv&query=%28%2A%29%20AND%20%28reviewed%3Atrue%29"```````## Joining blast table with annotation tableAt this point we have a blast output table and annotation table both with a Uniprot accession number. Thus we can join the two tables and be able to get more functional information about thet genes.```{bash}head -2 ../output/Ab_4-uniprot_blastx.tabwc -l ../output/Ab_4-uniprot_blastx.tab``````{bash}tr '|' '\t' < ../output/Ab_4-uniprot_blastx.tab | head -2``````{bash}tr '|' '\t' < ../output/Ab_4-uniprot_blastx.tab \> ../output/Ab_4-uniprot_blastx_sep.tab``````{bash}head -2 ../data/uniprot_table_r2023_01.tabwc -l ../data/uniprot_table_r2023_01.tab``````{r}library(tidyverse)library("kableExtra")``````{r}bltabl <-read.csv("../output/Ab_4-uniprot_blastx_sep.tab", sep ='\t', header =FALSE)``````{r, cache=TRUE}#spgo <- read.csv("../data/uniprot_table_r2023_01.tab", sep = '\t', header = TRUE)``````{r, echo=FALSE}#str(spgo)``````{r, eval = FALSE}kbl(head( left_join(bltabl, spgo, by = c("V3" = "Entry")) %>% select(V1, V3, V13, Protein.names, Organism, Gene.Ontology..biological.process., Gene.Ontology.IDs) %>% mutate(V1 = str_replace_all(V1, pattern = "solid0078_20110412_FRAG_BC_WHITE_WHITE_F3_QV_SE_trimmed", replacement = "Ab")))) %>% kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"))``````{r, eval = FALSE}left_join(bltabl, spgo, by = c("V3" = "Entry")) %>% select(V1, V3, V13, Protein.names, Organism, Gene.Ontology..biological.process., Gene.Ontology.IDs) %>% mutate(V1 = str_replace_all(V1, pattern = "solid0078_20110412_FRAG_BC_WHITE_WHITE_F3_QV_SE_trimmed", replacement = "Ab")) %>% write_delim("../output/blast_annot_go.tab", delim = '\t')```