
Determining exon and intron methylation
An effort to splice out exon and intron methylation levels on a per gene basis.
First thing needed was exon and intron beds that have gene ID information linked.
/home/shared/bedtools2/bin/intersectBed \
-wb \
-a ../genome-features/C_virginica-3.0_Gnomon_exon.bed \
-b ../genome-features/C_virginica-3.0_Gnomon_genes.bed \
| awk -v OFS="\t" '{ print $1, $2, $3, $7}' \
> ../genome-features/C_virginica-3.0_Gnomon_exon-geneID.bed
/home/shared/bedtools2/bin/intersectBed \
-wb \
-a ../genome-features/C_virginica-3.0_intron.bed \
-b ../genome-features/C_virginica-3.0_Gnomon_genes.bed \
| awk -v OFS="\t" '{ print $1, $2, $3, $7}' \
> ../genome-features/C_virginica-3.0_Gnomon_intron-geneID.bed
Then intersecting 10 bedgraphs
cd ../data/big/
FILES=$(ls *bedgraph)
cd -
for file in ${FILES}
do
NAME=$(echo ${file} | awk -F "_" '{print $1}')
echo ${NAME}
/home/shared/bedtools2/bin/intersectBed \
-wb \
-a ../data/big/${NAME}_R1_val_1_10x.bedgraph \
-b ../genome-features/C_virginica-3.0_Gnomon_exon-geneID.bed \
| awk -v name=$NAME -v OFS="\t" '{ print $0, name}' \
> ../output/43-exon-intron-methylation/${NAME}_mExon.out
done
cd ../data/big/
FILES=$(ls *bedgraph)
cd -
for file in ${FILES}
do
NAME=$(echo ${file} | awk -F "_" '{print $1}')
echo ${NAME}
/home/shared/bedtools2/bin/intersectBed \
-wb \
-a ../data/big/${NAME}_R1_val_1_10x.bedgraph \
-b ../genome-features/C_virginica-3.0_Gnomon_intron-geneID.bed \
| awk -v name=$NAME -v OFS="\t" '{ print $0, name}' \
> ../output/43-exon-intron-methylation/${NAME}_mIntron.out
done
and then smashing all together
cat ../output/43-exon-intron-methylation/*_mExon.out > ../output/43-exon-intron-methylation/exon-meth_all-samples.out
cat ../output/43-exon-intron-methylation/*_mIntron.out > ../output/43-exon-intron-methylation/intron-meth_all-samples.out
Then into tidyverse
exon_meth <- read.delim("../output/43-exon-intron-methylation/exon-meth_all-samples.out", header = FALSE)
intron_meth <- read.delim("../output/43-exon-intron-methylation/intron-meth_all-samples.out", header = FALSE)
summarizing by geneID
em <- exon_meth %>%
mutate(art = paste(V8, V9, sep = '_')) %>%
group_by(art) %>%
summarize(avg_meth = mean(V4))
int <- intron_meth %>%
mutate(art = paste(V8, V9, sep = '_')) %>%
group_by(art) %>%
summarize(avg_meth = mean(V4))
and joining by gene; separating out sample ID and sex.
exint <- inner_join(em, int, by = "art") %>%
separate(art, into = c("gene", "sample"), sep = "_") %>%
separate(sample, into = c("number", "sex"), sep = -1)
A plot of exon v intron methylation for every gene
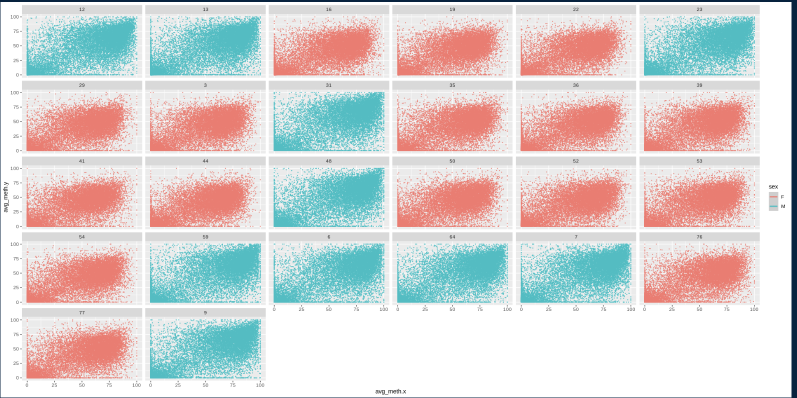
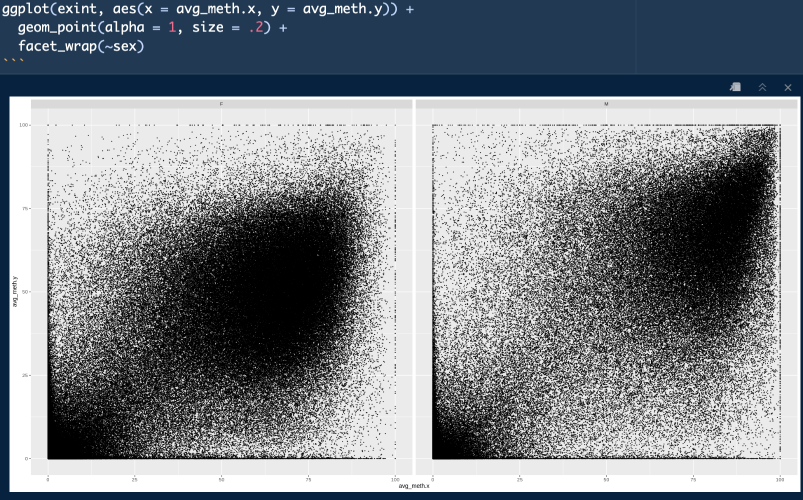
Seems to be interesting difference in sex
some csv files
https://github.com/sr320/ceabigr/tree/main/output/43-exon-intron-methylation
Written on October 23, 2022