
All the gigas in methylkit
Here want to take all the gigas - previously described and Bismarked - and see what we can glean from methylkit.
Run on Raven - code -> https://github.com/sr320/nb-2021/blob/main/C_gigas/code/60-allgig-methylkit.Rmd
wget -r \
--no-check-certificate \
--quiet \
--no-directories --no-parent \
-P /media/1TB_ext_SSD01/sr320/wd/ \
-A *sorted.bam \
https://gannet.fish.washington.edu/seashell/bu-mox/scrubbed/061021-big/
file.list_allgig=list('/media/1TB_ext_SSD01/sr320/wd/0501_R1_bismark_bt2_pe.deduplicated.sorted.bam',
'/media/1TB_ext_SSD01/sr320/wd/0502_R1_bismark_bt2_pe.deduplicated.sorted.bam',
'/media/1TB_ext_SSD01/sr320/wd/0503_R1_bismark_bt2_pe.deduplicated.sorted.bam',
'/media/1TB_ext_SSD01/sr320/wd/3501_R1_bismark_bt2_pe.deduplicated.sorted.bam',
...
)
myobj_all = processBismarkAln(location = file.list_allgig,
sample.id = list("0501","0502","0503","3501","3502","3503","5201","5202","5203","5901","5902","5903","zr3534_10","zr3534_1","zr3534_2","zr3534_3","zr3534_4","zr3534_5","zr3534_6","zr3534_7","zr3534_8","zr3534_9","zr3644_10","zr3644_11","zr3644_12","zr3644_13","zr3644_14","zr3644_15","zr3644_16","zr3644_17","zr3644_18","zr3644_19","zr3644_1","zr3644_20","zr3644_21","zr3644_22","zr3644_23","zr3644_24","zr3644_2","zr3644_3","zr3644_4","zr3644_5","zr3644_6","zr3644_7","zr3644_8","zr3644_9","zrtg3616_1","zrtg3616_2","zrtg3616_3","zrtg3616_4","zrtg3616_5","zrtg3616_6","zrtg3616_7","zrtg3616_8"),
assembly = "cv",
read.context="CpG",
mincov=2,
treatment = c(0,0,0,0,0,0,0,0,0,0,0,0,1,1,1,1,1,1,1,1,1,1,2,2,2,2,2,2,2,2,2,2,2,2,2,2,2,2,2,2,2,2,2,2,2,2,3,3,3,3,3,3,3,3))
filtered.myobj=filterByCoverage(myobj_all,lo.count=10,lo.perc=NULL,
hi.count=NULL,hi.perc=98)
meth_filter=unite(filtered.myobj, min.per.group=NULL, destrand=TRUE)
clusterSamples(meth_filter, dist="correlation", method="ward", plot=TRUE)
PCASamples(meth_filter)
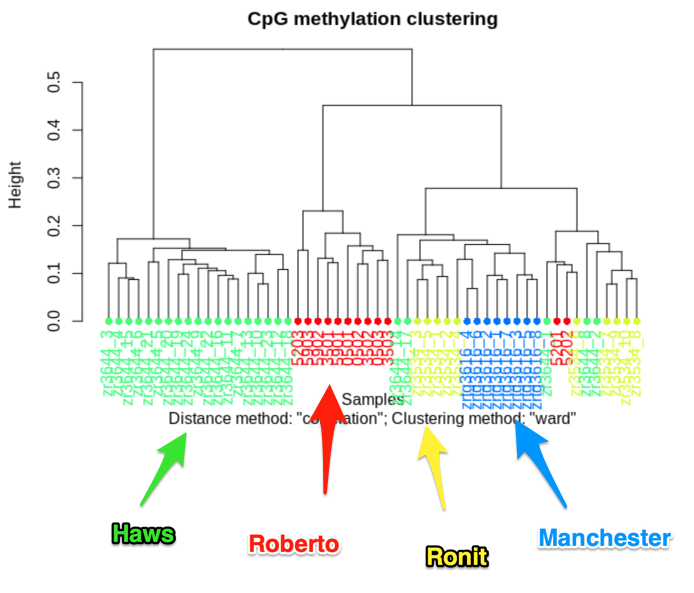
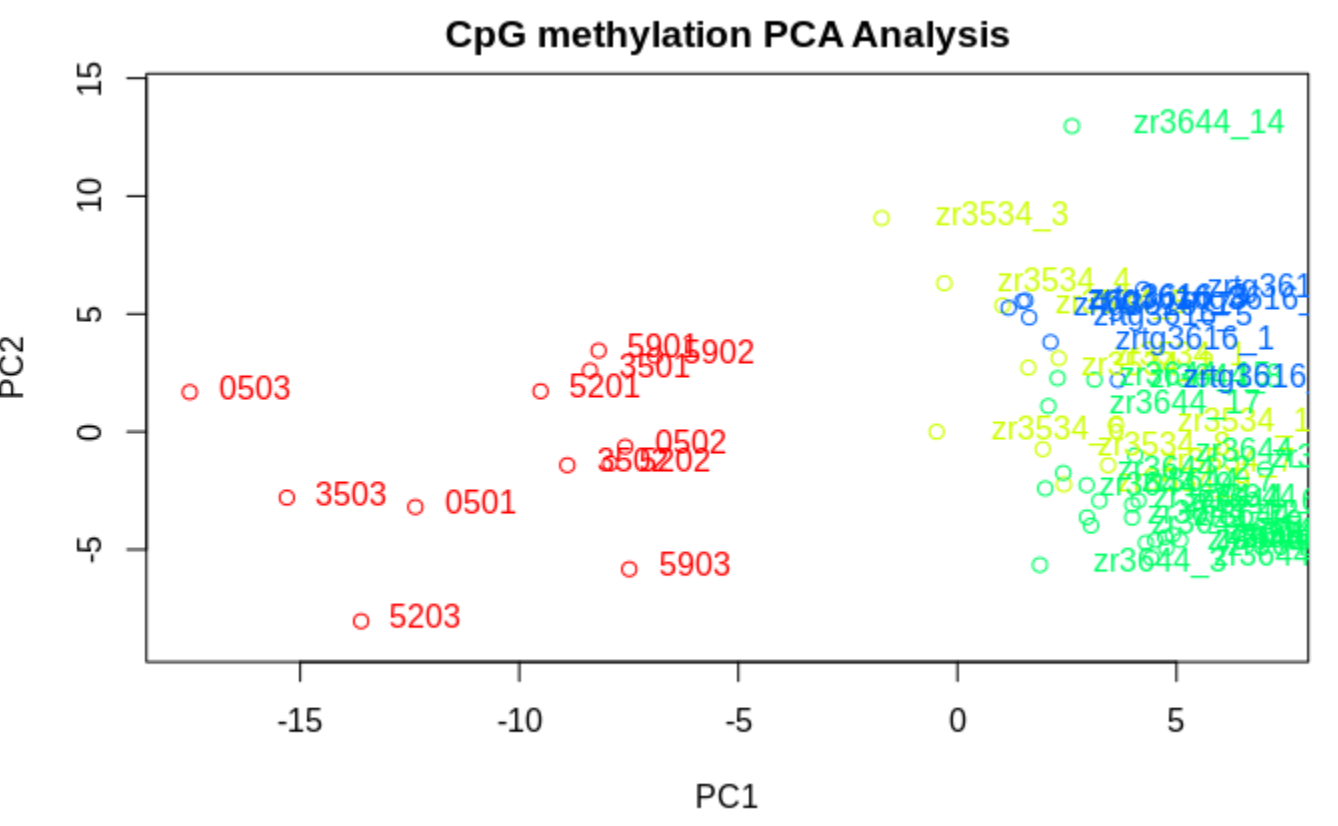
Running
myDiff=calculateDiffMeth(meth_filter,mc.cores=36)
and
# get hyper methylated bases
myDiff_a50p.hyper=getMethylDiff(myDiff,difference=50,qvalue=0.01,type="hyper")
#
# get hypo methylated bases
myDiff_a50p.hypo=getMethylDiff(myDiff,difference=50,qvalue=0.01,type="hypo")
#
#
# get all differentially methylated bases
myDiff_a50p=getMethylDiff(myDiff,difference=50,qvalue=0.01)
only gets 1 DML. Likely related to so few loci with coverage..
Written on November 15, 2021