Correlating the Matrices
56 - Matrix Synergy
Steven Roberts 11 September, 2023

The lab notebook of Steven Roberts
browse by date
Steven Roberts 11 September, 2023
Can we speed up samtools?
updated most recently August 18
Here is an attempt to pull relatedness / distance matrix from methylkit data…
Steven Roberts 11 August, 2023
link to rpub: https://rpubs.com/sr320/1070681
removing CT snps from CG methylation data.
code: https://github.com/mattgeorgephd/PSMFC-mytilus-byssus-pilot/blob/main/code/06-annotation.Rmd
Lets take look at Isoseq fasta
The following is a stepwise example or annotation of a gene set using UniProt::Swiss-Prot (reviewed) such that Gene Ontology terms can be associated with each gene.
In attempting to get quick comparison of alignment across genome, the question arises what is the difference (and accuracy) of kallisto (psuedo-align) and hisat. Spoiler - I was quite surprised with hisat w and w/o gtf. This is A pulcra RNA-seq data….
Initiating a look on how A pulcra will align to a few good genomes.
This is a running daily of all the stuff done in February and March. Or just some thoughts.
In preparation for lab meeting, some answer to Chris’s queries
This is a running daily of all the stuff done in January. Or just some thoughts.
My January goal is to try to make posting to my notebook easier. I would also like to get a better handle at project management.
This is a running daily of all the stuff done in November. Or just some thoughts
An effort to splice out exon and intron methylation levels on a per gene basis.
For a the ceabigr data lets ID which isoform is predominant, such that we can find out how treatment and/or methylation might influence this.
This is a running daily of all the stuff done in September. Or just some thoughts
Looking at number of cell, and expression data.
Here is some code for getting gene methylation. Will also add to handbook.
Some thoughts on the relationship of isoform count and methylation level.
This is a running daily of all the stuff done in August. Or just some thoughts
This is a running daily of all the stuff done in July.
This is a running daily of all the stuff done in June.
Video Recording from Prospective Student Days
Taking a deeper look at every step. Note this is single-end sequence data.
Here I want to examine how Machine Learning might compare with a our conventional gene expression analysis. The data set includes both male and female oysters exposed to OA conditions (and controls). Gonad tissue. Sam ran data through bowtie/stringtie for comparison. Complete sample details are below. PDF of post
With limited OA DMLs when considered in totality, looking within each sex to see what any OA influences might be. notebook: https://github.com/epigeneticstoocean/2018_L18-adult-methylation/blob/main/code/03.4-methylkit.Rmd
Digging into Cv DNA methylation data and I was trying to develop bedgraphs of libraries. I noted that in fact I did not have a complete set here. Having also recalled (and seen via .sh files) it took me at least 3 jobs to “complete” the effort that did not complete. So now I question everything. And disappointed that I failed to document the botch of an effort. Well today I am older and wiser and determined not to make a similar mistake. I have decided to cross my fingers and pull deduplicated bams back into mox and run downstream code. This of course presumes my bismark alignment and dedup was done properly.
Having previously taken a look at eastern oysters in OA to identify DMLs, here I attempt to take those data, redescribe and generate beds. TLDR: https://github.com/epigeneticstoocean/2018_L18-adult-methylation/tree/main/igv
Here want to take all the gigas - previously described and Bismarked - and see what we can glean from methylkit.
In an effort to couple DNA methylation data to complementary RNA-seq data we are looking at what the DNA methylation landscape, DML look like. Oysters were exposed to ocean acidification. Males and females were included.
02-Crab-qpcr
What if we start with current data and worked our way back to see if an integration of data was fruitful. Step 1 - bismark it all together..
Visiting the long ago Oly WBGS data. Will start with see if can simply reproduce.
After about a year away… here is something.
Video of Clam sampling: https://uw.hosted.panopto.com/Panopto/Pages/Viewer.aspx?id=8c080c5d-7e85-49f3-846b-ab9b012116db
Video of Clam sampling: https://uw.hosted.panopto.com/Panopto/Pages/Viewer.aspx?id=8c080c5d-7e85-49f3-846b-ab9b012116db
Going back to take a peek to see what treating geoduck EPI data as WGBS will look like.
We have some mapping done and will push forward with what we have his AM, which is a 100M subset of Mcap.
Step 1 is prepping 3 coral genomes
Last year :) I ran bismark on Oly samples that Laura is now digging into.
Updated: February 05
To have a general description of the geoduck methylation landscape, we take all 51 BS samples, concatenate, and map.
In preparation for the soon submission of the geoduck genome paper, I re-ran some mox jobs to see if we could reproduce the paper, particular with regard to the new naming scheme. The two scripts were 1217_1300.sh and 1218_1000.sh.
First prepared the genome
Went out to Pt Whitney with Shelly to semi-wrap up 40 day seed trial where parents experienced different OA regimes.
Taking the trimmed files @ /gscratch/srlab/strigg/data/Pgenr/FASTQS
and aligning to version 74 of the genome.
Working on the genetics / epigenetics paper, I decided to try to concatenate all reads and align with Bismark in order to get some basic stats.
To get a crude gene track for the Oly genome the big transcriptome was compared to genome.
We took DNA from a single Eastern oyster and prepped using MBD, MSP digestion, and plain old DNA. Full details can be found here.
I have to share some information with several new folks, and there are likely old folks that should give this a refresh… thus I am posting it here.
In an effort to improve an assembly here is a compilation of MP libraries with longest insert sizes. Specifically this would be an insert size of 8-10kb
Working in Rmd, here is getting some DMLs into IGV format. TLDR - bedfile
Full running of the OAKL samples.
As a better assembly is coming online for the Geoduck, we have started to look at Bismark mapping of prior samples.
In an attempt to start to visualize differences while waiting on hardware I brought some Bismark alignments into methyKit.
I have been working through Bismark with a few Crassosstrea virginica datasets. This includes the BS data from the 2015 Oil exposure experiment, OA exposure - gonad tissue (OAKL), and a full suite of library preps via Qiagen.
It seems that mapping rates can very a lot. We have a new data set comparing WG-BS, RRBS, and MBDBS. This is valuable as it offers data to run the math on what is most optimal. This first step is mapping. Here I explore CLC results as we are working with Qiagen and this is the software the are using.
Here are data corresponding to different types of library preparation.
I am exploring a few versions of the PBjelly Olympia oyster genome assembly and bisulfite read mapping.
There are several options for fasta output of Supernova assemblies.
Supernova completed the 10x Chromium data assembly.
About a week ago I started a Supernova 2.0 run to get some of this Chromium 10x data assembled.
We had a nice chat about how to reboot a couple of proteomic projects.
Running some alignments for Charlie.
As part of this Geoduck Larvae Trial we will be running some filters through proteomics and metagenomics.
Getting back to the command line.
Replacing toaster drive.
We have the following several Oly draft assemblies. Available here
Running QUAST to compare genome assembly.
Here is a Summary of the Illumina NS++ data dump (by platform).
Some summary environmental data for DNR project
Exploring the different aspects of data generated for the Oly genome.
Per this issue I took at look at some of Yaamini’s data.
Sam has compiled the current status of Olympia oyster genome assemblies here. I am going to try to assess differences.
We have a few MiSeq files.
Playing around a bit with Mox (crippled by lack of disk space). Ran FastQC
Here is a summary of the new data dump.
While we received a lot of files in HiSeq folder, none were fastq, thus Sam downloaded from BaseSpace. And it is ugly.
Using the Trandsdecoder and the Trinity assembly, a deduced proteome was generated.
Deduced protein sequences for 0804_Pgen_larvae.fasta
Having spent a day in Hyak, I think I know have a workflow that makes sense.
In preparation for new proteomic analysis here is a transcriptome from the NovaSeq.
Running Trinity on Mox. Geoduck larvae.
Running Trinity on EMU
Topic: Proteomic talk w/ Emma Date : Aug 3, 2017 10:53 AM Pacific Time (US and Canada)
Exploring RNA-Seq data from Illumina effort
Here is a set of videos where I
1) download annotations from UniProt
2) upload said file to the new SQLShare
3) upload Blastx output to SQLShare and…
4) do a left join.
Here is how one might go about visualizing Proteomic Data. This is based on a list of proteins Laura found to be different in geoducks in eel grass (as opposed to not being in eel grass).
comp138254
comp142216
comp125530
comp48421
comp144401
comp135856
comp143411
comp122035
comp134625
comp144270
comp142142
comp143197
comp143411
comp142396
comp88705
comp144180
comp131660
comp128586
comp28288
comp144604
comp141473
comp139766
comp116351
comp129221
comp22527
comp134200
comp136492
comp133552
comp144504
comp141096
comp99434
comp142358
comp143236
comp124813
comp144421
comp131211
comp143770
comp144132
comp127542
comp133562
comp142424
comp142890
comp135129
comp134692
comp144262
comp143418
comp133063
comp144191
comp90334
comp139531
comp142589
comp137055
comp143502
comp131651
comp141946
comp139881
comp143082
comp130569
comp143835
comp153529
comp128923
comp114823
comp143766
comp142589
comp135181
comp137628
comp140039
comp144637
comp137991
comp123956
comp128513
comp144581
comp135366
comp141512
Exploring various options for comparative genomic in CoGe
Yesterday Emma was concerned about Rhonda’s Abacus file. If fact there were differences. I created a new Abacus parameter file
In an attempt to go from mzXML to Abacus, I took Rhonda’s mzXML files on Emu, copied them to my directory and rand the following
Here is an attempt to annotate about half of the 41 DMRs Sean has identified.
Sean has identified 41 loci that are different in the 3 treatments at Day 10!
Having run the first batch of geoduck RRBS throught CoGe - Here is the mCpG file and information of how these files were generated.
Fifty RRBS Libraries were constructed by Hollie and sequenced (Maybe? these numbers do not match nightingales).
I went out to Manchester yesterday and checked on the TripleT (Two Treatment Trial) project.
Another quarter is complete for our Bioinformatics class , and once again we learned a bit.
!find /Volumes/web/nightingales/O_lurida/20160223_gbs/1NF*1.fq.gz | xargs basename -s _1.fq.gz \
| xargs -I{} /Applications/bioinfo/bowtie2-2.2.4/bowtie2 \
-x /Users/sr320/git-repos/student-fish546-2016/data/Ostrea_lurida-Scaff-10k-bowtie-index \
-1 /Volumes/web/nightingales/O_lurida/20160223_gbs/{}_1.fq.gz \
-2 /Volumes/web/nightingales/O_lurida/20160223_gbs/{}_2.fq.gz \
-p 8 \
--very-sensitive-local \
-S /Volumes/caviar/wd/2016-12-01/{}.sam
``` D-128-95-149-192:~ sr320$ brew install coreutils Updating Homebrew… ==> Auto-updated Homebrew! Updated 1 tap (homebrew/core). ==> Updated Formulae mercurial
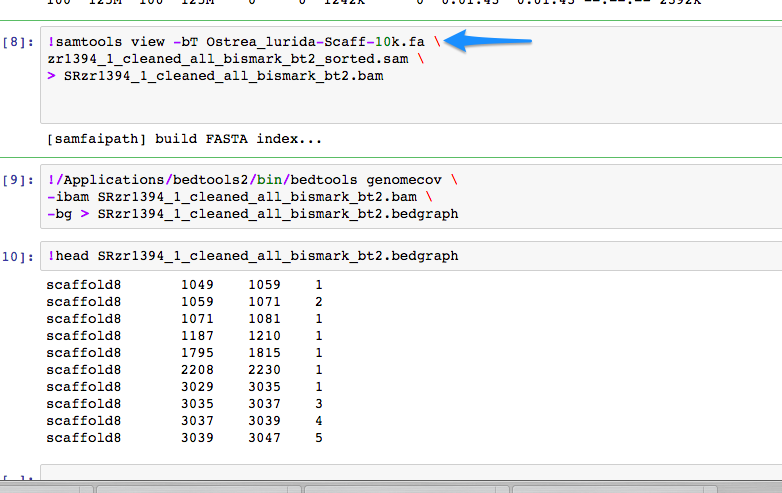
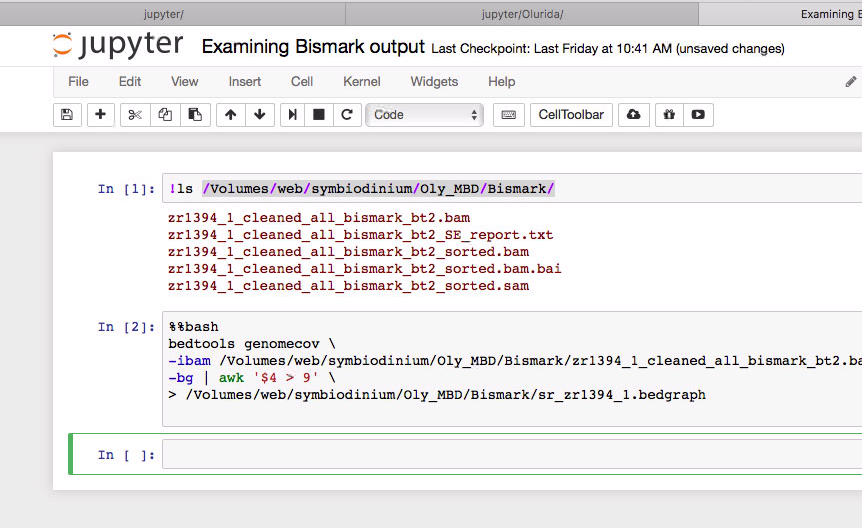
Mapped RNA-seq reads yesterday. Today trying 2bRAD that matches BS data.
!/Applications/bioinfo/bowtie2-2.2.4/bowtie2 \
-x ../data/Ostrea_lurida-Scaff-10k-bowtie-index \
-1 /Volumes/web/nightingales/O_lurida/filtered_106A_Male_Mix_TAGCTT_L004_R1.fastq.gz \
-2 /Volumes/web/nightingales/O_lurida/filtered_106A_Male_Mix_TAGCTT_L004_R2.fastq.gz \
-p 6 \
--very-fast \
-S /Volumes/caviar/wd/2016-11-11/bw-106A_Male_Mix_TAGCTT_L004.sam
!samtools view -bS /Volumes/caviar/wd/2016-11-11/bw-106A_Male_Mix_TAGCTT_L004.sam \
| samtools sort -o /Volumes/caviar/wd/2016-11-11/bw-106A_Male_Mix_TAGCTT_L004.bam
!samtools index /Volumes/caviar/wd/2016-11-11/bw-106A_Male_Mix_TAGCTT_L004.bam
Ran Blastp against UniProt.
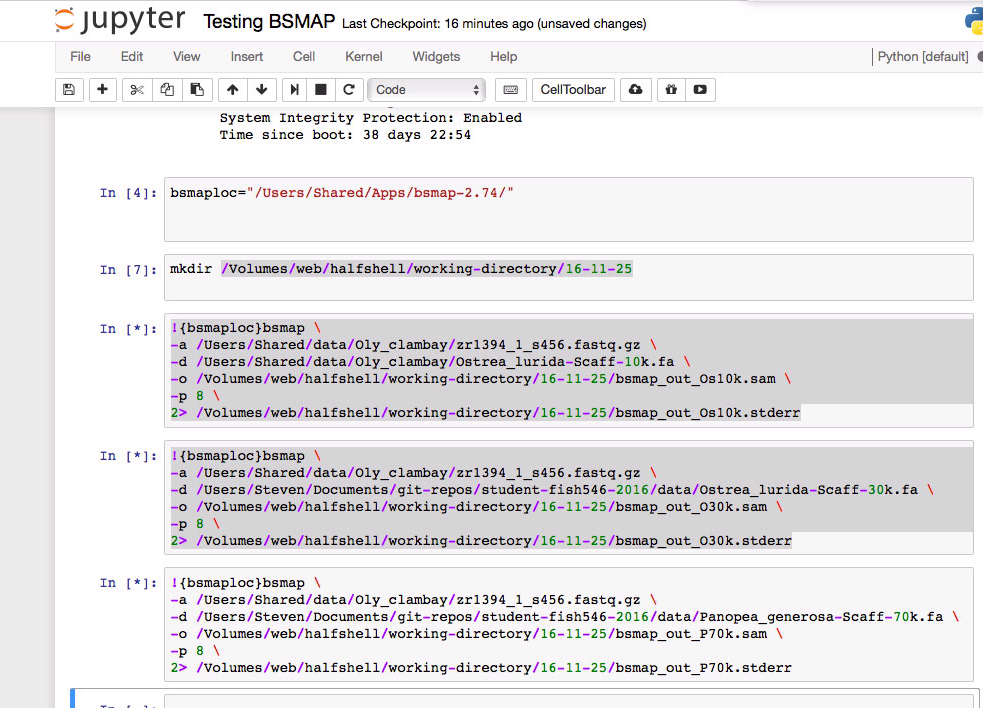
Running BSMAP on version of Ostrea lurida genome that is limited by 10k minimum scaffold threshold.
With a third blasting and comparing hits for all 10 parts of the query, I am satisfied with the output.
On genefish. Will try splitting.
Working on the finalizing the big table for the the transcriptome.
Link to notebook exploring the table https://github.com/sr320/paper-pano-go/blob/master/jupyter-nbs/11-Exploring-the-Big-Table.ipynb

Analysis of eight Fidalgo Olympia oysters. Maybe I just need a second sentence.
ls analyses/2016-10-11
mkfmt_M2.txt mkfmt_M3.txt
ls -lh /Volumes/caviar/wd/2016-10-11/bsmap*sam
-rw-r--r-- 1 sr320 staff 208M Oct 15 02:52 /Volumes/caviar/wd/2016-10-11/bsmap_out_1_ATCACG.sam
-rw-r--r-- 1 sr320 staff 254M Oct 16 04:21 /Volumes/caviar/wd/2016-10-11/bsmap_out_2_CGATGT.sam
-rw-r--r-- 1 sr320 staff 253M Oct 17 05:33 /Volumes/caviar/wd/2016-10-11/bsmap_out_3_TTAGGC.sam
-rw-r--r-- 1 sr320 staff 253M Oct 18 08:19 /Volumes/caviar/wd/2016-10-11/bsmap_out_4_TGACCA.sam
-rw-r--r-- 1 sr320 staff 264M Oct 20 15:50 /Volumes/caviar/wd/2016-10-11/bsmap_out_5_ACAGTG.sam
-rw-rw-rw- 1 sr320 staff 263M Oct 22 02:37 /Volumes/caviar/wd/2016-10-11/bsmap_out_6_GCCAAT.sam
-rw-rw-rw- 1 sr320 staff 225M Oct 20 18:49 /Volumes/caviar/wd/2016-10-11/bsmap_out_7_CAGATC.sam
-rw-rw-rw- 1 sr320 staff 299M Oct 19 18:55 /Volumes/caviar/wd/2016-10-11/bsmap_out_8_ACTTGA.sam
-rw-r--r-- 1 sr320 staff 1.5G Oct 11 08:06 /Volumes/caviar/wd/2016-10-11/bsmap_out_M2.sam
-rw-r--r-- 1 sr320 staff 1.6G Oct 11 08:10 /Volumes/caviar/wd/2016-10-11/bsmap_out_M3.sam
bsmaploc="/Applications/bioinfo/BSMAP/bsmap-2.74/"
cd /Volumes/caviar/wd/2016-10-11/
/Volumes/caviar/wd/2016-10-11
for i in ("1_ATCACG","2_CGATGT","3_TTAGGC","4_TGACCA","5_ACAGTG","6_GCCAAT","7_CAGATC","8_ACTTGA"):
!python {bsmaploc}methratio.py \
-d ../data/Ostrea_lurida.scafSeq \
-u -z -g \
-o methratio_out_{i}.txt \
-s {bsmaploc}samtools \
bsmap_out_{i}.sam \
@ Sat Oct 22 09:46:37 2016: reading reference ../data/Ostrea_lurida.scafSeq ...
@ Sat Oct 22 09:47:26 2016: reading bsmap_out_1_ATCACG.sam ...
[samopen] SAM header is present: 765755 sequences.
@ Sat Oct 22 09:47:53 2016: combining CpG methylation from both strands ...
@ Sat Oct 22 09:49:11 2016: writing methratio_out_1_ATCACG.txt ...
@ Sat Oct 22 09:54:10 2016: done.
total 467574 valid mappings, 618824 covered cytosines, average coverage: 2.02 fold.
@ Sat Oct 22 09:54:15 2016: reading reference ../data/Ostrea_lurida.scafSeq ...
@ Sat Oct 22 09:55:04 2016: reading bsmap_out_2_CGATGT.sam ...
[samopen] SAM header is present: 765755 sequences.
@ Sat Oct 22 09:55:37 2016: combining CpG methylation from both strands ...
@ Sat Oct 22 09:56:55 2016: writing methratio_out_2_CGATGT.txt ...
@ Sat Oct 22 10:01:55 2016: done.
total 579365 valid mappings, 689492 covered cytosines, average coverage: 2.19 fold.
@ Sat Oct 22 10:02:00 2016: reading reference ../data/Ostrea_lurida.scafSeq ...
@ Sat Oct 22 10:02:49 2016: reading bsmap_out_3_TTAGGC.sam ...
[samopen] SAM header is present: 765755 sequences.
@ Sat Oct 22 10:03:21 2016: combining CpG methylation from both strands ...
@ Sat Oct 22 10:04:39 2016: writing methratio_out_3_TTAGGC.txt ...
@ Sat Oct 22 10:09:37 2016: done.
total 579579 valid mappings, 678634 covered cytosines, average coverage: 2.24 fold.
@ Sat Oct 22 10:09:42 2016: reading reference ../data/Ostrea_lurida.scafSeq ...
@ Sat Oct 22 10:10:31 2016: reading bsmap_out_4_TGACCA.sam ...
[samopen] SAM header is present: 765755 sequences.
@ Sat Oct 22 10:11:04 2016: combining CpG methylation from both strands ...
@ Sat Oct 22 10:12:21 2016: writing methratio_out_4_TGACCA.txt ...
@ Sat Oct 22 10:17:20 2016: done.
total 577435 valid mappings, 690889 covered cytosines, average coverage: 2.18 fold.
@ Sat Oct 22 10:17:25 2016: reading reference ../data/Ostrea_lurida.scafSeq ...
@ Sat Oct 22 10:18:14 2016: reading bsmap_out_5_ACAGTG.sam ...
[samopen] SAM header is present: 765755 sequences.
@ Sat Oct 22 10:18:47 2016: combining CpG methylation from both strands ...
@ Sat Oct 22 10:20:04 2016: writing methratio_out_5_ACAGTG.txt ...
@ Sat Oct 22 10:25:04 2016: done.
total 608092 valid mappings, 691864 covered cytosines, average coverage: 2.27 fold.
@ Sat Oct 22 10:25:09 2016: reading reference ../data/Ostrea_lurida.scafSeq ...
@ Sat Oct 22 10:25:58 2016: reading bsmap_out_6_GCCAAT.sam ...
[samopen] SAM header is present: 765755 sequences.
@ Sat Oct 22 10:26:32 2016: combining CpG methylation from both strands ...
@ Sat Oct 22 10:27:51 2016: writing methratio_out_6_GCCAAT.txt ...
@ Sat Oct 22 10:32:57 2016: done.
total 604365 valid mappings, 689831 covered cytosines, average coverage: 2.27 fold.
@ Sat Oct 22 10:33:02 2016: reading reference ../data/Ostrea_lurida.scafSeq ...
@ Sat Oct 22 10:33:51 2016: reading bsmap_out_7_CAGATC.sam ...
[samopen] SAM header is present: 765755 sequences.
@ Sat Oct 22 10:34:20 2016: combining CpG methylation from both strands ...
@ Sat Oct 22 10:35:33 2016: writing methratio_out_7_CAGATC.txt ...
@ Sat Oct 22 10:40:32 2016: done.
total 507109 valid mappings, 646374 covered cytosines, average coverage: 2.09 fold.
@ Sat Oct 22 10:40:38 2016: reading reference ../data/Ostrea_lurida.scafSeq ...
@ Sat Oct 22 10:41:27 2016: reading bsmap_out_8_ACTTGA.sam ...
[samopen] SAM header is present: 765755 sequences.
@ Sat Oct 22 10:42:05 2016: combining CpG methylation from both strands ...
@ Sat Oct 22 10:43:22 2016: writing methratio_out_8_ACTTGA.txt ...
@ Sat Oct 22 10:48:21 2016: done.
total 689625 valid mappings, 732123 covered cytosines, average coverage: 2.42 fold.
#first methratio files are converted to filter for CG context, 3x coverage (mr3x.awk), and reformatting (mr_gg.awk.sh).
#due to issue passing variable to awk, simple scripts were used (included in repository)
for i in ("1_ATCACG","2_CGATGT","3_TTAGGC","4_TGACCA","5_ACAGTG","6_GCCAAT","7_CAGATC","8_ACTTGA"):
!echo {i}
!grep "[A-Z][A-Z]CG[A-Z]" <methratio_out_{i}.txt> methratio_out_{i}CG.txt
!awk -f /Users/sr320/git-repos/sr320.github.io/jupyter/scripts/mr3x.awk methratio_out_{i}CG.txt \
> mr3x.{i}.txt
!awk -f /Users/sr320/git-repos/sr320.github.io/jupyter/scripts/mr_gg.awk.sh \
mr3x.{i}.txt > mkfmt_{i}.txt
1_ATCACG
2_CGATGT
3_TTAGGC
4_TGACCA
5_ACAGTG
6_GCCAAT
7_CAGATC
8_ACTTGA
#maybe we need to ignore case
!md5 mkfmt_M2.txt mkfmti_M2.txt | head
MD5 (mkfmt_M2.txt) = df67fde9e87ec165618d384374074057
MD5 (mkfmti_M2.txt) = df67fde9e87ec165618d384374074057
#nope
!head -5 mkfmt*
==> mkfmt_1_ATCACG.txt <==
chr.Base chr base strand coverage freqC freqT
scaffold1.33 scaffold1 33 F 3 0.00 100.00
scaffold1.143 scaffold1 143 F 4 0.00 100.00
scaffold1.244 scaffold1 244 F 3 66.67 33.33
scaffold1.265 scaffold1 265 F 7 14.29 85.71
==> mkfmt_2_CGATGT.txt <==
chr.Base chr base strand coverage freqC freqT
scaffold1.33 scaffold1 33 F 11 0.00 100.00
scaffold1.143 scaffold1 143 F 9 0.00 100.00
scaffold1.566 scaffold1 566 F 8 0.00 100.00
scaffold1.572 scaffold1 572 F 3 0.00 100.00
==> mkfmt_3_TTAGGC.txt <==
chr.Base chr base strand coverage freqC freqT
scaffold1.12 scaffold1 12 F 4 25.00 75.00
scaffold1.33 scaffold1 33 F 3 0.00 100.00
scaffold1.109 scaffold1 109 F 5 0.00 100.00
scaffold1.143 scaffold1 143 F 9 0.00 100.00
==> mkfmt_4_TGACCA.txt <==
chr.Base chr base strand coverage freqC freqT
scaffold1.109 scaffold1 109 F 9 11.11 88.89
scaffold1.143 scaffold1 143 F 11 9.09 90.91
scaffold1.244 scaffold1 244 F 3 0.00 100.00
scaffold1.265 scaffold1 265 F 4 25.00 75.00
==> mkfmt_5_ACAGTG.txt <==
chr.Base chr base strand coverage freqC freqT
scaffold1.33 scaffold1 33 F 8 0.00 100.00
scaffold1.109 scaffold1 109 F 5 0.00 100.00
scaffold1.143 scaffold1 143 F 5 0.00 100.00
scaffold1.244 scaffold1 244 F 6 33.33 66.67
==> mkfmt_6_GCCAAT.txt <==
chr.Base chr base strand coverage freqC freqT
scaffold1.12 scaffold1 12 F 3 0.00 100.00
scaffold1.33 scaffold1 33 F 11 9.09 90.91
scaffold1.109 scaffold1 109 F 7 0.00 100.00
scaffold1.143 scaffold1 143 F 11 0.00 100.00
==> mkfmt_7_CAGATC.txt <==
chr.Base chr base strand coverage freqC freqT
scaffold1.33 scaffold1 33 F 10 0.00 100.00
scaffold1.109 scaffold1 109 F 6 0.00 100.00
scaffold1.143 scaffold1 143 F 16 0.00 100.00
scaffold1.244 scaffold1 244 F 3 0.00 100.00
==> mkfmt_8_ACTTGA.txt <==
chr.Base chr base strand coverage freqC freqT
scaffold1.33 scaffold1 33 F 7 0.00 100.00
scaffold1.109 scaffold1 109 F 4 0.00 100.00
scaffold1.143 scaffold1 143 F 10 10.00 90.00
scaffold1.244 scaffold1 244 F 6 0.00 100.00
==> mkfmt_M2.txt <==
chr.Base chr base strand coverage freqC freqT
scaffold1.14274 scaffold1 14274 F 4 0.00 100.00
scaffold1.14305 scaffold1 14305 F 4 0.00 100.00
scaffold1.15309 scaffold1 15309 F 4 0.00 100.00
scaffold1.15315 scaffold1 15315 F 4 0.00 100.00
==> mkfmt_M3.txt <==
chr.Base chr base strand coverage freqC freqT
scaffold1.259 scaffold1 259 F 4 100.00 0.00
scaffold1.263 scaffold1 263 F 4 100.00 0.00
scaffold1.267 scaffold1 267 F 4 100.00 0.00
scaffold1.271 scaffold1 271 F 4 100.00 0.00
==> mkfmti_M2.txt <==
chr.Base chr base strand coverage freqC freqT
scaffold1.14274 scaffold1 14274 F 4 0.00 100.00
scaffold1.14305 scaffold1 14305 F 4 0.00 100.00
scaffold1.15309 scaffold1 15309 F 4 0.00 100.00
scaffold1.15315 scaffold1 15315 F 4 0.00 100.00
==> mkfmti_M3.txt <==
chr.Base chr base strand coverage freqC freqT
scaffold1.259 scaffold1 259 F 4 100.00 0.00
scaffold1.263 scaffold1 263 F 4 100.00 0.00
scaffold1.267 scaffold1 267 F 4 100.00 0.00
scaffold1.271 scaffold1 271 F 4 100.00 0.00
cd git-repos/sr320.github.io/jupyter/
/Users/sr320/git-repos/sr320.github.io/jupyter
ls
[34mCgigas[m[m/ [34mOlurida[m[m/ [34manalyses[m[m/ [34mscripts[m[m/
mkdir analyses/$(date +%F)
for i in ("1_ATCACG","2_CGATGT","3_TTAGGC","4_TGACCA","5_ACAGTG","6_GCCAAT","7_CAGATC","8_ACTTGA"):
!cp /Volumes/caviar/wd/2016-10-11/mkfmt_{i}.txt analyses/$(date +%F)/mkfmt_{i}.txt
!head analyses/$(date +%F)/*
==> analyses/2016-10-22/mkfmt_1_ATCACG.txt <==
chr.Base chr base strand coverage freqC freqT
scaffold1.33 scaffold1 33 F 3 0.00 100.00
scaffold1.143 scaffold1 143 F 4 0.00 100.00
scaffold1.244 scaffold1 244 F 3 66.67 33.33
scaffold1.265 scaffold1 265 F 7 14.29 85.71
scaffold1.579 scaffold1 579 F 4 0.00 100.00
scaffold1.591 scaffold1 591 F 4 0.00 100.00
scaffold1.622 scaffold1 622 F 4 0.00 100.00
scaffold1.641 scaffold1 641 F 3 66.67 33.33
scaffold1.723 scaffold1 723 F 3 0.00 100.00
==> analyses/2016-10-22/mkfmt_2_CGATGT.txt <==
chr.Base chr base strand coverage freqC freqT
scaffold1.33 scaffold1 33 F 11 0.00 100.00
scaffold1.143 scaffold1 143 F 9 0.00 100.00
scaffold1.566 scaffold1 566 F 8 0.00 100.00
scaffold1.572 scaffold1 572 F 3 0.00 100.00
scaffold1.576 scaffold1 576 F 9 0.00 100.00
scaffold1.579 scaffold1 579 F 8 0.00 100.00
scaffold1.582 scaffold1 582 F 6 83.33 16.67
scaffold1.591 scaffold1 591 F 6 0.00 100.00
scaffold1.602 scaffold1 602 F 5 0.00 100.00
==> analyses/2016-10-22/mkfmt_3_TTAGGC.txt <==
chr.Base chr base strand coverage freqC freqT
scaffold1.12 scaffold1 12 F 4 25.00 75.00
scaffold1.33 scaffold1 33 F 3 0.00 100.00
scaffold1.109 scaffold1 109 F 5 0.00 100.00
scaffold1.143 scaffold1 143 F 9 0.00 100.00
scaffold1.244 scaffold1 244 F 4 0.00 100.00
scaffold1.265 scaffold1 265 F 11 0.00 100.00
scaffold1.566 scaffold1 566 F 5 0.00 100.00
scaffold1.576 scaffold1 576 F 5 0.00 100.00
scaffold1.579 scaffold1 579 F 6 0.00 100.00
==> analyses/2016-10-22/mkfmt_4_TGACCA.txt <==
chr.Base chr base strand coverage freqC freqT
scaffold1.109 scaffold1 109 F 9 11.11 88.89
scaffold1.143 scaffold1 143 F 11 9.09 90.91
scaffold1.244 scaffold1 244 F 3 0.00 100.00
scaffold1.265 scaffold1 265 F 4 25.00 75.00
scaffold1.566 scaffold1 566 F 4 0.00 100.00
scaffold1.572 scaffold1 572 F 3 0.00 100.00
scaffold1.576 scaffold1 576 F 7 0.00 100.00
scaffold1.579 scaffold1 579 F 5 0.00 100.00
scaffold1.582 scaffold1 582 F 4 50.00 50.00
==> analyses/2016-10-22/mkfmt_5_ACAGTG.txt <==
chr.Base chr base strand coverage freqC freqT
scaffold1.33 scaffold1 33 F 8 0.00 100.00
scaffold1.109 scaffold1 109 F 5 0.00 100.00
scaffold1.143 scaffold1 143 F 5 0.00 100.00
scaffold1.244 scaffold1 244 F 6 33.33 66.67
scaffold1.265 scaffold1 265 F 6 0.00 100.00
scaffold1.566 scaffold1 566 F 3 0.00 100.00
scaffold1.572 scaffold1 572 F 3 0.00 100.00
scaffold1.576 scaffold1 576 F 4 0.00 100.00
scaffold1.579 scaffold1 579 F 5 0.00 100.00
==> analyses/2016-10-22/mkfmt_6_GCCAAT.txt <==
chr.Base chr base strand coverage freqC freqT
scaffold1.12 scaffold1 12 F 3 0.00 100.00
scaffold1.33 scaffold1 33 F 11 9.09 90.91
scaffold1.109 scaffold1 109 F 7 0.00 100.00
scaffold1.143 scaffold1 143 F 11 0.00 100.00
scaffold1.244 scaffold1 244 F 9 11.11 88.89
scaffold1.265 scaffold1 265 F 11 0.00 100.00
scaffold1.566 scaffold1 566 F 10 0.00 100.00
scaffold1.572 scaffold1 572 F 4 0.00 100.00
scaffold1.576 scaffold1 576 F 11 0.00 100.00
==> analyses/2016-10-22/mkfmt_7_CAGATC.txt <==
chr.Base chr base strand coverage freqC freqT
scaffold1.33 scaffold1 33 F 10 0.00 100.00
scaffold1.109 scaffold1 109 F 6 0.00 100.00
scaffold1.143 scaffold1 143 F 16 0.00 100.00
scaffold1.244 scaffold1 244 F 3 0.00 100.00
scaffold1.265 scaffold1 265 F 9 0.00 100.00
scaffold1.566 scaffold1 566 F 6 0.00 100.00
scaffold1.576 scaffold1 576 F 5 0.00 100.00
scaffold1.579 scaffold1 579 F 6 16.67 83.33
scaffold1.582 scaffold1 582 F 5 80.00 20.00
==> analyses/2016-10-22/mkfmt_8_ACTTGA.txt <==
chr.Base chr base strand coverage freqC freqT
scaffold1.33 scaffold1 33 F 7 0.00 100.00
scaffold1.109 scaffold1 109 F 4 0.00 100.00
scaffold1.143 scaffold1 143 F 10 10.00 90.00
scaffold1.244 scaffold1 244 F 6 0.00 100.00
scaffold1.265 scaffold1 265 F 7 14.29 85.71
scaffold1.566 scaffold1 566 F 7 0.00 100.00
scaffold1.576 scaffold1 576 F 6 0.00 100.00
scaffold1.579 scaffold1 579 F 7 14.29 85.71
scaffold1.582 scaffold1 582 F 7 42.86 57.14
url for 8 tables..
https://github.com/sr320/sr320.github.io/tree/master/jupyter/analyses/2016-10-22
Last year Cris sent me some Atlantic salmon lncRNAs (~21k) where he wanted to know what the adjacent gene ID.
Reposted from the FISH546 Project
I started analysis of two gigas samples to eventually be compared with methylRAD. Below is a snapshot of the Jupyter notebook.
Updating @ https://github.com/sr320/sr320.github.io/blob/master/jupyter/Cgigas/Lotterhos%20BS%20samples.ipynb
The M2 and M3 samples are here:
http://owl.fish.washington.edu/nightingales/C_gigas/9_GATCAG_L001_R1_001.fastq.gz http://owl.fish.washington.edu/nightingales/C_gigas/10_TAGCTT_L001_R1_001.fastq.gz
bsmaploc="/Applications/bioinfo/BSMAP/bsmap-2.74/"
!curl \
ftp://ftp.ensemblgenomes.org/pub/release-32/metazoa/fasta/crassostrea_gigas/dna/Crassostrea_gigas.GCA_000297895.1.dna_sm.toplevel.fa.gz \
> /Volumes/caviar/wd/data/Crassostrea_gigas.GCAz_000297895.1.dna_sm.toplevel.fa.gz
% Total % Received % Xferd Average Speed Time Time Time Current
Dload Upload Total Spent Left Speed
100 148M 100 148M 0 0 5192k 0 0:00:29 0:00:29 --:--:-- 5790k
!curl ftp://ftp.ensemblgenomes.org/pub/release-32/metazoa/fasta/crassostrea_gigas/dna/CHECKSUMS
08778 148199 Crassostrea_gigas.GCA_000297895.1.dna.nonchromosomal.fa.gz
08778 148199 Crassostrea_gigas.GCA_000297895.1.dna.toplevel.fa.gz
57175 143732 Crassostrea_gigas.GCA_000297895.1.dna_rm.nonchromosomal.fa.gz
57175 143732 Crassostrea_gigas.GCA_000297895.1.dna_rm.toplevel.fa.gz
45604 151782 Crassostrea_gigas.GCA_000297895.1.dna_sm.nonchromosomal.fa.gz
45604 151782 Crassostrea_gigas.GCA_000297895.1.dna_sm.toplevel.fa.gz
62118 5 README
!ls /Volumes/caviar/wd/data/
Crassostrea_gigas.GCAz_000297895.1.dna_sm.toplevel.fa.gz
!md5 /Volumes/caviar/wd/data/Crassostrea_gigas.GCAz_000297895.1.dna_sm.toplevel.fa.gz
MD5 (/Volumes/caviar/wd/data/Crassostrea_gigas.GCAz_000297895.1.dna_sm.toplevel.fa.gz) = c70084d76bd6d7a1ba52c13843e69ccc
cd /Volumes/caviar/wd/
/Volumes/caviar/wd
mkdir $(date +%F)
ls
[34m2016-10-11[m[m/ [34mdata[m[m/
ls /Volumes/web/nightingales/C
!curl \
http://owl.fish.washington.edu/nightingales/C_gigas/9_GATCAG_L001_R1_001.fastq.gz \
> /Volumes/caviar/wd/2016-10-11/9_GATCAG_L001_R1_001.fastq.gz
% Total % Received % Xferd Average Speed Time Time Time Current
Dload Upload Total Spent Left Speed
100 560M 100 560M 0 0 55.6M 0 0:00:10 0:00:10 --:--:-- 77.8M
!curl \
http://owl.fish.washington.edu/nightingales/C_gigas/10_TAGCTT_L001_R1_001.fastq.gz \
> /Volumes/caviar/wd/2016-10-11/10_TAGCTT_L001_R1_001.fastq.gz
% Total % Received % Xferd Average Speed Time Time Time Current
Dload Upload Total Spent Left Speed
100 619M 100 619M 0 0 46.1M 0 0:00:13 0:00:13 --:--:-- 44.0M
cd 2016-10-11/
/Volumes/caviar/wd/2016-10-11
!cp 9_GATCAG_L001_R1_001.fastq.gz M2.fastq.gz
!cp 10_TAGCTT_L001_R1_001.fastq.gz M3.fastq.gz
for i in ("M2","M3"):
!{bsmaploc}bsmap \
-a {i}.fastq.gz \
-d ../data/Crassostrea_gigas.GCAz_000297895.1.dna_sm.toplevel.fa \
-o bsmap_out_{i}.sam \
-p 6
BSMAP v2.74
Start at: Tue Oct 11 08:02:27 2016
Input reference file: ../data/Crassostrea_gigas.GCAz_000297895.1.dna_sm.toplevel.fa (format: FASTA)
Load in 7658 db seqs, total size 557717710 bp. 8 secs passed
total_kmers: 43046721
Create seed table. 24 secs passed
max number of mismatches: read_length * 8% max gap size: 0
kmer cut-off ratio: 5e-07
max multi-hits: 100 max Ns: 5 seed size: 16 index interval: 4
quality cutoff: 0 base quality char: '!'
min fragment size:28 max fragemt size:500
start from read #1 end at read #4294967295
additional alignment: T in reads => C in reference
mapping strand: ++,-+
Single-end alignment(6 threads)
Input read file: M2.fastq.gz (format: gzipped FASTQ)
Output file: bsmap_out_M2.sam (format: SAM)
Thread #1: 100000 reads finished. 30 secs passed
Thread #0: 50000 reads finished. 30 secs passed
Thread #2: 150000 reads finished. 31 secs passed
Thread #3: 200000 reads finished. 31 secs passed
Thread #5: 250000 reads finished. 31 secs passed
Thread #4: 300000 reads finished. 31 secs passed
Thread #1: 350000 reads finished. 36 secs passed
Thread #0: 400000 reads finished. 36 secs passed
Thread #2: 450000 reads finished. 36 secs passed
Thread #3: 500000 reads finished. 36 secs passed
Thread #5: 550000 reads finished. 37 secs passed
Thread #4: 600000 reads finished. 37 secs passed
Thread #1: 650000 reads finished. 42 secs passed
Thread #2: 750000 reads finished. 42 secs passed
Thread #0: 700000 reads finished. 42 secs passed
Thread #3: 800000 reads finished. 42 secs passed
Thread #5: 850000 reads finished. 42 secs passed
Thread #4: 900000 reads finished. 43 secs passed
Thread #1: 950000 reads finished. 48 secs passed
Thread #2: 1000000 reads finished. 48 secs passed
Thread #3: 1100000 reads finished. 48 secs passed
Thread #0: 1050000 reads finished. 49 secs passed
Thread #5: 1150000 reads finished. 49 secs passed
Thread #4: 1200000 reads finished. 49 secs passed
Thread #1: 1250000 reads finished. 54 secs passed
Thread #2: 1300000 reads finished. 54 secs passed
Thread #3: 1350000 reads finished. 55 secs passed
Thread #5: 1450000 reads finished. 55 secs passed
Thread #4: 1500000 reads finished. 55 secs passed
Thread #0: 1400000 reads finished. 55 secs passed
Thread #1: 1550000 reads finished. 60 secs passed
Thread #2: 1600000 reads finished. 60 secs passed
Thread #3: 1650000 reads finished. 61 secs passed
Thread #4: 1750000 reads finished. 61 secs passed
Thread #5: 1700000 reads finished. 61 secs passed
Thread #0: 1800000 reads finished. 61 secs passed
Thread #1: 1850000 reads finished. 67 secs passed
Thread #2: 1900000 reads finished. 67 secs passed
Thread #3: 1950000 reads finished. 68 secs passed
Thread #4: 2000000 reads finished. 68 secs passed
Thread #5: 2050000 reads finished. 68 secs passed
Thread #0: 2100000 reads finished. 68 secs passed
Thread #1: 2150000 reads finished. 73 secs passed
Thread #2: 2200000 reads finished. 74 secs passed
Thread #3: 2250000 reads finished. 74 secs passed
Thread #4: 2300000 reads finished. 74 secs passed
Thread #5: 2350000 reads finished. 74 secs passed
Thread #0: 2400000 reads finished. 75 secs passed
Thread #1: 2450000 reads finished. 80 secs passed
Thread #2: 2500000 reads finished. 80 secs passed
Thread #3: 2550000 reads finished. 80 secs passed
Thread #4: 2600000 reads finished. 81 secs passed
Thread #5: 2650000 reads finished. 81 secs passed
Thread #0: 2700000 reads finished. 81 secs passed
Thread #2: 2800000 reads finished. 86 secs passed
Thread #1: 2750000 reads finished. 86 secs passed
Thread #3: 2850000 reads finished. 86 secs passed
Thread #4: 2900000 reads finished. 87 secs passed
Thread #5: 2950000 reads finished. 87 secs passed
Thread #0: 3000000 reads finished. 88 secs passed
Thread #2: 3050000 reads finished. 92 secs passed
Thread #1: 3100000 reads finished. 92 secs passed
Thread #3: 3150000 reads finished. 92 secs passed
Thread #4: 3200000 reads finished. 92 secs passed
Thread #5: 3250000 reads finished. 93 secs passed
Thread #0: 3300000 reads finished. 94 secs passed
Thread #2: 3350000 reads finished. 98 secs passed
Thread #1: 3400000 reads finished. 98 secs passed
Thread #3: 3450000 reads finished. 98 secs passed
Thread #4: 3500000 reads finished. 98 secs passed
Thread #5: 3550000 reads finished. 99 secs passed
Thread #0: 3600000 reads finished. 100 secs passed
Thread #2: 3650000 reads finished. 104 secs passed
Thread #1: 3700000 reads finished. 104 secs passed
Thread #3: 3750000 reads finished. 104 secs passed
Thread #4: 3800000 reads finished. 104 secs passed
Thread #5: 3850000 reads finished. 105 secs passed
Thread #0: 3900000 reads finished. 106 secs passed
Thread #2: 3950000 reads finished. 110 secs passed
Thread #1: 4000000 reads finished. 110 secs passed
Thread #3: 4050000 reads finished. 110 secs passed
Thread #4: 4100000 reads finished. 110 secs passed
Thread #5: 4150000 reads finished. 111 secs passed
Thread #0: 4200000 reads finished. 112 secs passed
Thread #2: 4250000 reads finished. 116 secs passed
Thread #1: 4300000 reads finished. 116 secs passed
Thread #3: 4350000 reads finished. 116 secs passed
Thread #4: 4400000 reads finished. 117 secs passed
Thread #5: 4450000 reads finished. 117 secs passed
Thread #0: 4500000 reads finished. 119 secs passed
Thread #2: 4550000 reads finished. 122 secs passed
Thread #1: 4600000 reads finished. 122 secs passed
Thread #3: 4650000 reads finished. 122 secs passed
Thread #4: 4700000 reads finished. 123 secs passed
Thread #5: 4750000 reads finished. 123 secs passed
Thread #0: 4800000 reads finished. 125 secs passed
Thread #2: 4850000 reads finished. 128 secs passed
Thread #1: 4900000 reads finished. 128 secs passed
Thread #3: 4950000 reads finished. 129 secs passed
Thread #4: 5000000 reads finished. 129 secs passed
Thread #5: 5050000 reads finished. 129 secs passed
Thread #0: 5100000 reads finished. 131 secs passed
Thread #2: 5150000 reads finished. 134 secs passed
Thread #1: 5200000 reads finished. 134 secs passed
Thread #3: 5250000 reads finished. 134 secs passed
Thread #4: 5300000 reads finished. 135 secs passed
Thread #5: 5350000 reads finished. 135 secs passed
Thread #0: 5400000 reads finished. 137 secs passed
Thread #2: 5450000 reads finished. 140 secs passed
Thread #1: 5500000 reads finished. 140 secs passed
Thread #3: 5550000 reads finished. 141 secs passed
Thread #4: 5600000 reads finished. 141 secs passed
Thread #5: 5650000 reads finished. 141 secs passed
Thread #0: 5700000 reads finished. 143 secs passed
Thread #2: 5750000 reads finished. 147 secs passed
Thread #1: 5800000 reads finished. 147 secs passed
Thread #3: 5850000 reads finished. 147 secs passed
Thread #4: 5900000 reads finished. 147 secs passed
Thread #5: 5950000 reads finished. 148 secs passed
Thread #0: 6000000 reads finished. 150 secs passed
Thread #2: 6050000 reads finished. 153 secs passed
Thread #1: 6100000 reads finished. 153 secs passed
Thread #3: 6150000 reads finished. 153 secs passed
Thread #4: 6200000 reads finished. 153 secs passed
Thread #5: 6250000 reads finished. 154 secs passed
Thread #0: 6300000 reads finished. 156 secs passed
Thread #1: 6400000 reads finished. 160 secs passed
Thread #2: 6350000 reads finished. 160 secs passed
Thread #4: 6500000 reads finished. 160 secs passed
Thread #3: 6450000 reads finished. 160 secs passed
Thread #5: 6550000 reads finished. 161 secs passed
Thread #0: 6600000 reads finished. 164 secs passed
Thread #1: 6650000 reads finished. 166 secs passed
Thread #4: 6750000 reads finished. 167 secs passed
Thread #2: 6700000 reads finished. 167 secs passed
Thread #3: 6800000 reads finished. 167 secs passed
Thread #5: 6850000 reads finished. 168 secs passed
Thread #0: 6900000 reads finished. 171 secs passed
Thread #1: 6950000 reads finished. 173 secs passed
Thread #2: 7050000 reads finished. 174 secs passed
Thread #4: 7000000 reads finished. 174 secs passed
Thread #3: 7100000 reads finished. 174 secs passed
Thread #5: 7150000 reads finished. 174 secs passed
Thread #0: 7200000 reads finished. 177 secs passed
Thread #1: 7250000 reads finished. 179 secs passed
Thread #2: 7300000 reads finished. 180 secs passed
Thread #4: 7350000 reads finished. 180 secs passed
Thread #3: 7400000 reads finished. 180 secs passed
Thread #5: 7450000 reads finished. 180 secs passed
Thread #0: 7500000 reads finished. 184 secs passed
Thread #1: 7550000 reads finished. 186 secs passed
Thread #2: 7600000 reads finished. 186 secs passed
Thread #4: 7650000 reads finished. 187 secs passed
Thread #3: 7700000 reads finished. 187 secs passed
Thread #5: 7750000 reads finished. 187 secs passed
Thread #0: 7800000 reads finished. 191 secs passed
Thread #1: 7850000 reads finished. 193 secs passed
Thread #2: 7900000 reads finished. 193 secs passed
Thread #4: 7950000 reads finished. 193 secs passed
Thread #3: 8000000 reads finished. 193 secs passed
Thread #5: 8050000 reads finished. 193 secs passed
Thread #0: 8100000 reads finished. 196 secs passed
Thread #1: 8150000 reads finished. 198 secs passed
Thread #2: 8200000 reads finished. 199 secs passed
Thread #4: 8250000 reads finished. 199 secs passed
Thread #3: 8300000 reads finished. 199 secs passed
Thread #5: 8350000 reads finished. 199 secs passed
Thread #0: 8400000 reads finished. 203 secs passed
Thread #1: 8450000 reads finished. 205 secs passed
Thread #2: 8500000 reads finished. 205 secs passed
Thread #4: 8550000 reads finished. 205 secs passed
Thread #5: 8650000 reads finished. 205 secs passed
Thread #3: 8600000 reads finished. 205 secs passed
Thread #0: 8700000 reads finished. 209 secs passed
Thread #1: 8750000 reads finished. 210 secs passed
Thread #2: 8800000 reads finished. 211 secs passed
Thread #4: 8850000 reads finished. 211 secs passed
Thread #5: 8900000 reads finished. 211 secs passed
Thread #3: 8950000 reads finished. 211 secs passed
Thread #0: 9000000 reads finished. 215 secs passed
Thread #1: 9050000 reads finished. 216 secs passed
Thread #2: 9100000 reads finished. 217 secs passed
Thread #4: 9150000 reads finished. 217 secs passed
Thread #5: 9200000 reads finished. 217 secs passed
Thread #3: 9250000 reads finished. 217 secs passed
Thread #0: 9300000 reads finished. 221 secs passed
Thread #1: 9350000 reads finished. 222 secs passed
Thread #2: 9400000 reads finished. 223 secs passed
Thread #4: 9450000 reads finished. 223 secs passed
Thread #5: 9500000 reads finished. 223 secs passed
Thread #3: 9550000 reads finished. 223 secs passed
Thread #0: 9600000 reads finished. 227 secs passed
Thread #1: 9650000 reads finished. 228 secs passed
Thread #2: 9700000 reads finished. 228 secs passed
Thread #4: 9750000 reads finished. 229 secs passed
Thread #5: 9800000 reads finished. 229 secs passed
Thread #3: 9850000 reads finished. 229 secs passed
Thread #0: 9900000 reads finished. 233 secs passed
Thread #1: 9950000 reads finished. 234 secs passed
Thread #2: 10000000 reads finished. 235 secs passed
Thread #4: 10050000 reads finished. 235 secs passed
Thread #5: 10100000 reads finished. 235 secs passed
Thread #3: 10150000 reads finished. 235 secs passed
Thread #0: 10200000 reads finished. 239 secs passed
Thread #1: 10250000 reads finished. 240 secs passed
Thread #2: 10300000 reads finished. 241 secs passed
Thread #4: 10350000 reads finished. 241 secs passed
Thread #5: 10400000 reads finished. 241 secs passed
Thread #3: 10450000 reads finished. 241 secs passed
Thread #2: 10564512 reads finished. 242 secs passed
for i in ("M2","M3"):
!python {bsmaploc}methratio.py \
-d ../data/Crassostrea_gigas.GCAz_000297895.1.dna_sm.toplevel.fa \
-u -z -g \
-o methratio_out_{i}.txt \
-s {bsmaploc}samtools \
bsmap_out_{i}.sam \
We sampled 96 oysters that were part of Katherine Silliman’s summer project. These oysters were from three locales and had spent about 48 hours in OA treatment (half in contol water). Full sensor data is available here.
Getting closer to a master table for a the gonad transcriptome.
After kicking around how to make a very big table with all of the annotations… I finally made some progress.
Curious to see how Jay might tackle genome assembly (and looking ahead to FISH546) I wanted to see what could be done. I was able to bring an SRA file directly into Cyverse
Frustrated with roll you own option in with EBI GO association files etc. I am trialing Blast2GO commandline. It was not much better getting going but it is downloading stuff now.
As per this pipeline I will run the 8 individuals in the environmental epigenetics mini-experiment.